Each one of these is about the size of a bowl of soup. In some places the springs are so numerous that it is difficult to avoid stepping in them. You can tell just by looking at these three springs that the chemistry varies considerably; I’m given to understand that the different colors are due to the dominant oxidation species of sulfur, and the one on the far left was about thirty degrees hotter than the other two. All three of them are almost certainly colonized by fascinating microbes.
The experienced microbiologists on the expedition set about the business of pursuing questions like Who is there? and What are they doing? I was there to collect a few samples for metagenomic sequencing, and so my own work was completed on the first day. I spent the rest of my time there thinking about the microbes that live in these beautiful hotsprings, and wondering How did they get there?
Extremophiles are practically made-to-order for this question. The study of extremophile biology has been a bonanza for both applied and basic science. Extremophiles live differently, and their adaptations have taught us a lot about how evolution works, about the history of life on earth, about biochemistry, and all sorts of interesting things. However, their very peculiarity poses an interesting problem. Imagine you would freeze to death at 80° Celsius. How does the world look to you? Pretty inhospitable; a few little ponds of warmth dotted across vast deserts of freezing death.
Ecologists have known for a very long time that these barriers an
d conduits are crucial evolutionary mechanisms. Evolution can be seen as an interaction of two processes; mutation and selection. The nature of the interaction is determined by the structure of the population in which they occur. This structure is determined by biological processes such as sexual mechanisms and recombination, which are in turn is determined chiefly by the population’s distribution in space and its migration in that space.
As any sports fan knows, the structure of a tournament can be more important than the outcome of any particular game, or even the rules of the game. This is true for life, too. From one generation to the next, genes are shuffled and reshuffled through the population, and the way the population is compartmentalized sets the broad outlines of this process.
A monolithic population — one in which all players are in the same compartment — evolves differently than a fragmented population, even if mutation, recombination and selection pressures are identical. And so, if we want to understand the evolution of microbes, we need to know something about this structure. Bass Becking’s hypothesis is a statement about the nature of this structure, specifically, that the structure is monolithic. If true, it means that the only difference between an Erlenmeyer flask and the entire planet is the number of unique niches. The difference in size would be irrelevant.
This is a pretty strange thing to claim. And yet, the Baas Becking model has proved surprisingly difficult to knock down. For as long as microbiologists have been systematically classifying microbes, whenever they’ve found similar environments, they’ve found basically the same microbes. Baas Becking proposed his hypothesis in an environment of overwhelming evidence.
However, as molecular techniques have allowed researchers to probe deeper into the life and times of microbes (and every other living thing), some cracks have started to show. Rachel Whitaker and Thane Papke have challenged the Bass Becking model by looking at the biogeography of thermophilic microbes (such as Sulfolobus islandicus and Oscillatoria amphigranulata), first by 16S rRNA phylogenetics and later using high resolution, multi-locus methods. Both Rachel’s work and Papke’s work, as well as many studies of disease evolution, very clearly show that when you look within a microbial species, the populations do not appear quite so cosmopolitan. While Sulfolobus islandicus is found in hot springs all over the world, the evolutionary distance between each pair of its isolates is strongly correlated with the geographic distance between their sources. So, these microbes are indeed getting around the planet, but if we look at their DNA, we see that they are not getting around so quickly.
However, Baas Becking has an answer for this; “
…but the environment selects.” What if the variation is due to selection acting at a finer scale? It’s
well established that
species sorting effects play a major role in determining the composition of microbial communities at the species level. There is no particular reason to believe that this effect does not apply at smaller phylogenetic scales. The work with
Sulfolobus islandicus attempts to control for this by choosing isolates from hot springs with similar physical and chemical properties, but unfortunately there is no such thing as a pair of identical hot springs. Just walk the boardwalks in Yellowstone, and you’ll see what I mean. The differences among the sites from which these microbes were isolated can always be offered as an alternative explanation to dispersal. Even if you crank those differences down to nearly zero, one can always suggest that perhaps there is a difference that we don’t know about that happened to be important.
This is why the Baas Becking hypothesis is so hard to refute: One must simultaneously establish that there is a non-uniform phylogeographic distribution, and that this non-uniformity is not due to selection-driven effects such as species sorting or local adaptive selection. To do this, we need a methodology that allows us to simultaneously measure phylogeography and selection.
There are a variety of ways of measuring selection. Jonathan’s Evolution textbook has a whole chapter about it. I’ll go into a bit more detail in Aim 3, but for now, I’d just like to draw attention to the fact that the effect of selection does not typically fall uniformly across a genome. This non-uniformity tends to leave a characteristic signature in the nucleotide composition of a population. Selective sweeps and bottlenecks, for example, are usually identified by examining how a population’s nucleotide diversity varies over its genome.
For certain measures of selection (e.g., linkage disequilibrium) one can design a set of marker genes that could be used to assay the relative effect of selection among populations. This could then extend the single species, multi-locus phylogenetic methods that have already been used to measure the biogeography of microbes to include information about selection. This could, in principle, allow one to simultaneously refute “everything is everywhere…” and “…but the environment selects.” However, designing and testing all those markers, ordering all those primers and doing all those PCR reactions would be a drag. If selection turned out to work a little differently than initially imagined, the data would be useless.
But, these are microbes, after all. If I’ve learned anything from Jonathan, it’s that there is very little to be gained by avoiding sequencing.
We’re getting better and better at sequencing new genomes, but it is not a trivial undertaking. However, re-sequencing genomes is becoming routine enough it’s replacing microarray analysis for many applications. The most difficult part of re-sequencing an isolate is growing the isolate. Fortunately, re-sequencing is particularly well suited for culture-independent approaches. As long as we have complete genomes for the organisms we’re interested in, we can build metagenomes from environmental samples using our favorite second-generation sequencing platform. Then we simply map the reads to the reference genomes. The workflow is a bit like ChIP-seq, except without culturing anything and without the ChIP. We go directly from the environmental sample to sequencing to read-mapping. Maybe we can call it Eco-seq? That sounds catchy.
Not only is the whole-genome approach better, but with the right tools, it is easier and cheaper that multi-locus methods, and allows one to include many species simultaneously. The data will do beautifully for phylogeography, and have the added benefit that we can recapitulate the multi-locus methodology by throwing away data, rather collecting more.
To implement this, I have divided my project into three main steps :
- Aim 1 : Develop a biogeographical sampling strategy to optimize representation of a natural microbial community
- Aim 2 : Develop an apply techniques for broad matagenomic sampling, metadata collection and data processing
- Aim 3 : Test the dispersal hypothesis using a phylogeographic model with controls for local selection
But, before I get into the implementation, I should pause for a moment and make sure I’ve stated my hypothesis perfectly clearly : I think that dispersal plays a major role in the composition of microbial communities. The Baas Becking hypothesis doesn’t deny that dispersal happens, in fact, it asserts that dispersal is infinite, but that it is selection, not dispersal, that ultimately determines which microbes are found in any particular place. If I find instead that dispersal itself plays a major role in determining community composition, then the world is a very different place to be a microbe.
Aim 1 : Develop a biogeographical sampling strategy to optimize the representation of a complete natural community
While I would love to keep visiting places like Kamchatka and Yellowstone, I’ve decided to study the biogeography of halophiles, specifically in California and neighboring states. Firstly, because I can drive and hike to most of the places were they grow. Secondly, because the places where halophiles like to grow tend to be much easier to get permission to sample from. Some of them are industrial waste sites; no worry about disturbing fragile habitats. Thirdly, because our lab has been heavily involved in sequencing halophile genomes, which are necessary component of my approach. There is also a fourth reason, but I’m saving it for the Epilogue.
As I have
written about before, the US Geological Survey has built
a massive catalog of hydrological features across the Western United States. It’s as complete a list of the substantial, persistent halophile habitats one could possibly wish for. It has almost two thousand possible sites in California, Nevada and Oregon alone :

USGS survey sites. UC Davis is marked with a red star.
The database is complete enough that we can get a pretty good sense of what the distribution of sites looks like within this region just by looking at the map. The sites are basically coincident with mountain ranges. Even though they aren’t depicted, the Coastal Range, the Sierras, the Cascades and the Rockies all stand out. This isn’t surprising; salt lakes require some sort of constraining geographic topology, or the natural drainage would simply carry the salt into the ocean. Interestingly, hot springs are also usually found in mountains (some of these sites are indeed hot springs), but that has less to do with the mountains themselves as it does with the processes that built mountains. To put it more pithily, you find salt lakes where there are mountains, but you find mountains where there are hot springs.
This database obviously contains too many sites to visit. It took Dr. Mariner’s team forty years to gather all of this information. I need to choose from among these sites. But which ones? Is there a way to know if I’m making good selections? Does it even matter?
As it turns out,
it does matter. When we talk about dispersal in the context of biogeography, we are making a statement about the way organisms get from place to place. Usually, we expect to see a
distance decay relationship, because we expect that more distant places are harder to get to, and thus the rates of dispersal across longer distances should be lower. I need to be reasonably confident that I will see the same distance-decay relationship within the sub-sample that I would have seen for every site in the database. This doesn’t necessarily mean that the microbes will obey this relationship, but if they do, I need data that would support the measurement.
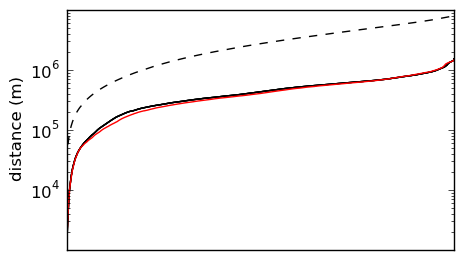
There is a pretty straightforward way of doing this. If we take every pair of sites in the database, calculate the Great Circle distance between them, and then sort these distances, we can get spectrum of pairwise distances. Here’s what that looks like for the sites in my chunk of the USGS database :
The spectrum of pairwise distances among all sites in the USGS databse (solid black), among randomly placed sites over the same geographic area (dashed black), and among random sub-sample of 360 sites from the database (solid red).
I’ve plotted three spectra here. The dashed black line is what you’d get if the sites had been randomly distributed over the same geographic area, and the solid black line is the spectra of the actual pairwise distances. As you can see, the distribution is highly non-random, but we already knew this just by glancing at the map. The red line is the spectrum of a random sub-sample of 360 sites from the database (I chose 360 because that is about how many samples I could collect in five one-week road trips).
This sub-sample matches the spectrum of the database pretty well, but not perfectly. It’s easy to generate candidate sub-samples, and they can be scored by how closely their spectra match the database. I’d like to minimize the amount of time it takes me to finish my dissertation, which I expect will be somewhat related to the number of samples I collect. There is a cute little optimization problem there.
Although I’ve outlined the field work, laboratory work and analysis as separate steps, these things will actually take place simultaneously. After I return from the field with the first batch of samples, I will process and submit them for sequencing before going on the next collection trip. I can dispatch the analysis pipeline from pretty much anywhere (even with my mobile phone). That’s why I’ve set aside sample selection and collection as a separate aim. The sample selection process determines where to start, how to proceed, and when I’m done.
Aim 2 : Develop an apply techniques for broad matagenomic sampling, metadata collection and data processing
In order to build all these genomes, I need to solve some technical problems. Building this many metagenomes is a pretty new thing, and so some of the tools I need did not exist in a form (or at a cost) that is useful to me. So, I’ve developed or adapted some new tools to bring the effort, cost and time for large-scale comparative metagenomics into the realm of a dissertation project.
There are four technical challenges :
- Quickly collect a large number of samples and transport them to the laboratory without degradation.
- Build several hundred sequencing libraries.
- Collect high-quality metadata describing the sites.
- Assemble thousands of re-sequenced genomes.
To solve each of these problems, I’ve applied exactly the same principle : Simplify and parallelize. I can’t claim credit for the idea here, because I was raised on it.
Literally.
Sample collection protocol
When I first joined Jonathan’s lab, Jenna Morgan (if you’re looking for her newer papers, make sure to add “Lang,” as she’s since gotten married) was testing how well metagenomic sequencing actually represents the target environment. In her paper,
now out in PLoS ONE, one of the key findings is that mechanical disruption is essential.
I learned during my trip to Kamchatka that getting samples back to the lab without degradation is very hard, and it really would be best to do the DNA extraction immediately. Unfortunately, another lesson I learned in Kamchatka is that it is surprisingly difficult to do molecular biology in the woods. One of the ways I helped out while I was there was to kill mosquitoes trying to bite our lab technician so she wouldn’t have to swat them with her gloved hands. It’s not easy to do this without making an aerosol of bug guts and blood over the open spin columns.
So, I was very excited when I went to ASM last year, and encountered a cool idea from Zymo Research. Basically, it’s a battery-operated bead mill, and a combined stabilization and cell lysis buffer. This solves the transportation problem and the bead-beating problem, without the need to do any fiddly pipetting and centrifuging in the field. Also, it looks cool.

Unfortunately, the nylon screw threads on the sample processor tend to get gummed up with dirt, so I’ve designed my own attachment that uses a quick-release style fitting instead of a screw top.
It’s called the Smash-o-Tron 3000, and you can download it on Thingiverse.
Sequencing library construction
The next technical problem is actually building the sequencing libraries. Potentially, there could be a lot of them, especially if I do replicates. If I were to collect three biological replicates from every site on the map, I would have to create about six thousand metagenomes. I will not be collecting anywhere close to six thousand samples, but I thought it was an interesting technical problem. So I solved it.
Well, actually I added some mechanization to a solution Epicentre (now part of Illumina) marketed, and my lab-mates Aaron Darling and Qingyi Zhang have refined into a dirt-cheap multiplexed sequencing solution. The standard technique for building Illumina sequencing libraries involves mechanically shearing the source DNA, ligating barcode sequences and sequencing adapters to the fragments, mixing them all together, and then doing size selection and cleanup. The first two steps of this process are fairly tedious and expensive. As it turns out,
Tn5 transposase can be used to fragment the DNA and ligate the barcodes and adapters in one easy digest. Qingyi is now growing huge quantities of the stuff.
The trouble is that DNA extraction yields an unpredictable amount of DNA, and the activity of Tn5 is sensitive to the concentration of target DNA. So, before you can start the Tn5 digest, you have to dilute the raw DNA to the right concentration and aliquat the correct amount for the reaction. This isn’t a big deal if you have a dozen samples. If you have thousands, the dilutions become the rate limiting step. If I’m the one doing the dilutions, it becomes a show-stopper at around a hundred samples. I’m just not that good at pipetting. (Seriously.)
The usual way of dealing with this problem is to use a liquid handling robot. Unfortunately, liquid handling robots are stupendously expensive. Even at their considerable expense, many of them are shockingly slow.
To efficiently process a large number of samples, we need to be able to treat every sample exactly the same. This way, can bang through the whole protocol with a multichannel pipetter. It occurred to me that many companies sell DNA extraction kits that use spin columns embedded in 96-well plates, and we have a swinging bucket centrifuge with a rotor that accommodates four plates at a time. So, the DNA extraction step is easy to parallelize. The Tn5 digests work just fine in 96-well plates.
We happen to have (well, actually
Marc’s lab has) a fluorometer that handles 96-well plates. Once the DNA extraction is finished, I can use a multichannel pipetter to make aliquats from the raw DNA, and measure the DNA yield for each sample in parallel. So far, so good.
Now, to dilute the raw DNA to the right concentration for the Tn5 digest, I need to put an equal volume of raw DNA into differing amounts of water. This violates the principle of treating every sample the same, which means I can’t use a multichannel pipetter to get the job done. That is, unless I have a 96-well plate that looks like this :
Programmatically generated dilution plate CAD model
I wrote a piece of software that takes a table of concentration measurements from the fluorometer, and designs a 96-well plate with wells of the correct volume to dilute each sample to the right concentration for the Tn5 digest. If I make one of these plates for each batch of 96 samples, I can use a multichannel pipetter throughout.
Of course, unless you are
Kevin Flynn, you can’t actually pipette liquids into a 3D computer model and achieve the desired effect. To convert the model from bits into atoms, I ordered a 3D printer kit from
Ultimaker. (I
love working in this lab!)
The Ultimaker kit
After three days of intense and highly entertaining fiddling around, I managed to get the kit assembled. A few more days of experimentation yielded my first successful prints (a couple of whistles). A few days after that, I was starting my first attempts to build my calibrated volume dilution plates.
Learning about 3D printing has been an adventure, but I’ve got the basics down and I’m now refining the process. I’m now printing plates with surprisingly good quality. I’ve had some help from the Ultimaker community on this, particularly from
Florian Horsch.
Much to my embarrassment, the first (very lousy) prototype of my calibrated volume dilution plate ended up on AggieTV. Fortunately, the glare from the window made it look much more awesome than it actual was.
The upshot is that if I needed to make ten or twenty thousand metagenomes, I could do it. I can print twelve 96-well dilution plates overnight. Working at a leisurely pace, these would allow me to make 1152 metagenome libraries in about two afternoons’ worth of work.
I’m pretty excited about this idea, and there are a lot of different directions one could take it. The College of Engineering here at UC Davis is letting me teach a class this quarter that I’ve decided to call “Robotics for Laboratory Applications,” where we’ll be exploring ways to apply this technology to molecular biology, genomics and ecology. Eight really bright UC Davis undergraduates have signed up (along with the director of the Genome Center’s Bioinformatics Core), and I’m very excited to see what they’ll do!
Environmental metadata collection
To help me sanity check the selection measurement, I decided that I wanted to have detailed measurements of environmental differences among sample sites. Water chemistry, temperature, weather, and variability of these are known to select for or against various species of microbes. The USGS database has extremely detailed measurements of all of these things, all the way down to the isotopic level. However, I still need to take my own measurements to confirm that the site hasn’t changed since it was visited by the USGS team, and to get some idea of what the variability of these parameters might be. It would also be nice if I could retrieve the data remotely, and not have to make return trips to every site.
Unfortunately, these products are are extraordinarily expensive. The ones that can be left in the field for a few months to log data cost even more. The ones that can transmit the data wirelessly are so expensive that I’d only be able to afford a handful if I blew an entire R01 grant on them.
This bothers me on a moral level. The key components are a few probes, a little lithium polymer battery, a solar panel the size of your hand, and a cell phone. You can buy them separately for maybe fifty bucks, plus the probes. Buying them as an integrated environmental data monitoring solution costs tens of thousands of dollars per unit. A nice one, with weather monitoring, backup batteries and a good enclosure could cost a hundred thousand dollars. You can make whatever apology you like on behalf of the industry, but the fact is that massive overcharging for simple electronics is preventing science from getting done.
So, I ordered a couple of Arduino boards and made my own.
My prototype Arduino-based environmental data logger. This version has a pH probe, Flash storage, and a Bluetooth interface.
The idea is to walk into the field with a data logger and a stick. Then I will find a suitable rock. Then I will pound the stick into the mud with the rock. Then I will strap the data logger to the stick, and leave it there while I go about the business of collecting samples. To keep it safe from the elements, the electronics will be entombed in a protective wad of silicone elastomer with a little solar panel and a battery.
The bill of materials for one of these data loggers is about $200, and so I won’t feel too bad about simply leaving them there to collect data. If the site has cell phone service, I will add a GSM modem to the datalogger (I like the
LinkSprite SM5100B with SparkFun’s GSM shield), and transmit the data to my server at UC Davis through an SMS gateway. Then I don’t have to go back to the site to collect the data. This could easily save $200 worth of gasoline. I’ll put a pre-paid return shipping labels on them so that they can find their way home someday. I’m eagerly looking forward to decades of calls from Jonathan complaining about my old grimy data loggers showing up in his mail.
From the water, the data logger can measure pH, dissolved oxygen, oxidation/reduction potential, conductivity (from which salinity can be calculated), and temperature. I may also add a small weather station to record air temperature, precipitation, wind speed and direction, and solar radiation. I doubt if all of these parameters will be useful, but the additional instrumentation is not very expensive.
Assembling the genomes
The final technical hurdle is assembling genomes from the metagenomic data. If I have 360 sites and 100 reference genomes, I’m going to have to assemble 36,000 genomes. Happily, I am really re-sequencing them, which is much, much easier than de novo sequencing. Nevertheless, 36,000 is still a lot of genomes.
For each metagenome, I must :
- Remove adapter contamination with TagDust
- Trim reads for quality, discard low quality reads
- Remove PCR duplicates
- Map reads to references with bwa, bowtie, SHRiMP, or whatever
This yields a
BAM file for each metagenome, each representing an alignment of reads to each scaffold of each reference genome. All of the reference genomes can be placed into a single FASTA file with a consistent naming scheme for distinguishing among scaffolds belonging to different organisms. A hundred-odd archaeal reference genomes is about 200-400 megabases, or an order of magnitude smaller than the human genome. Using the Burrows-Wheeler Aligner on a reasonably modern computer, this takes just a few minutes for each metagenome.
I’m impatient, though, and so I applied for (and received) an
AWS in Education grant. Then I wrote a script that parcels each metagenome off to a virtual machine image, and then unleashes all of them simultaneously on Amazon.com’s
thundering heard of rental computers. Once they finish their alignment, each virtual machine stores the BAM file in my
Dropbox account and shuts down. The going rate for an EC2 Extra Large instance is $0.68 per hour.
This approach could be used for any re-sequencing project, including ChIP-seq, RNA-seq, SNP analysis, and many others.
Aim 3 : Test the dispersal hypothesis using a phylogeographic model with controls for local selection
In order to test my hypothesis, I need to model the dispersal of organisms among the sites. However, in order to do a proper job of this, I need to make sure I’m not conflating dispersal and selective effects in the data used to initialize the model. There are three steps :
- Identify genomic regions that have recently been under selection
- Build genome trees with those regions masked out
- Model dispersal among the sites
In all three cases, there are a large number of methods to choose from.
One way of detecting the effects of selection is
Tajima’s D. This measures deviation from the
neutral model by comparing two estimators of the neutral genetic variation, one based on the nucleotide diversity and one based on the number of polymorphic sites. Neutral theory predicts that the two estimators are equal, and so genomic regions in which these two estimators are not equal are evolving in a way that is not predicted by the neutral model (i.e., they are under some kind of selection). One can do this calculation on a sliding window to measure Tajima’s
D for each coordinate of each the genome of each organism. As it turns out, th
is exact approach was used by David Begun’s lab to study the distribution of selection across the Drosophilia genome. I will delete the regions of the genomes that deviate significantly (say, by more than one standard deviation) from neutral. Then I’ll make whole genome alignments, and build a phylogenetic trees for each organism. This tree would contain only characters that (at least insofar as you believe Tajima’s D and Wu and Fey’s FST) are evolving neutrally, and are not under selection.
A phylogenetic tree represents evolutionary events that have taken place over time. In order to infer the dispersal of the represented organisms, would need model where those events took place. Again, there are a
variety of methods for doing this, and but my personal favorite is probably the approach used by
Isabel Sanmartín for modeling dispersal of invertebrates among the Canary Islands. I don’t know if this is necessarily the best method, but I like the idea that the DNA model and the dispersal model use the same mathematics, and are computed together. Basically, they allowed each taxa to evolve its own DNA model, but constrained by the requirement that they share a common dispersal model. Then they did Markov Chain Monte Carlo (MCMC) sampling of the posterior distributions of island model parameters (using
MrBayes 4.0).
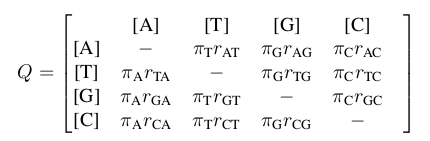
According to Wikipedia, the most respected and widely consulted authority on this and every topic, the
General Time Reversible Model it is the most generalized model describing the rates at which one nucleotide replaces another. If we want to know the rate at which a thymine turns into a guanine, we look at elment (2,3) of this matrix :
πG is the stationary state frequency for guanine, and rTG is the exchangability rate between from T to G. However, if we think of this a little differently, as Sanmartín suggests in her paper, we can use the GTR model for the dispersal of species among sites (or islands). If we want to know the rate at which a species migrates from island B to island C, we look in cell (2,3) of a very similar matrix :
Here, πC is the relative carrying capacity of island C, and rBC is the relative dispersal rate from island B to island C. Thus, the total dispersal from island i to island j is
dij = Nπirijπjm
where N is the total number of species in the system, and m is the group-specific dispersal rate. This might look something like this :
One nifty thing I discovered about MrBayes is that it can link against the
BEAGLE library, which can accelerate these calculations using GPU clusters. Suspiciously,
Aaron Darling is
one of the authors. If you were looking for evidence that the Eisen Lab is a den of Bayesians, this would be it.
This brings us, at last, back to the hypothesis and Baas Becking. Here we have a phylogeographic model of dispersal among sites within a metacommunity, with the effects of selection removed. If the model predicts well-supported finite rates of dispersal within the metacommunity, my hypothesis is sustained. If not, then Baas Becking’s 78 year reign continues.
Epilogue : Lourens Baas Becking, the man verses the strawman
Lourens Baas Becking
Microbiologists have been taking potshots at the Baas Becking hypothesis for a decade or two now, and I am no exception. I’m certainly hoping that the study I’ve outlined here will be the fatal blow.
However, it’s important to recognize that we’ve been a bit unfair to Baas Becking himself. The hypothesis that carries his name is a
model, and Baas Becking himself fully understood that dispersal must play an important role in community formation. He understood perfectly well that “
alles is overal: maar het milieu selecteert” was not literally true; it is only mostly true, and then only in the context of the observational methodology available at the time. In 1934, in the same book where he proposed his eponymous hypothesis, he observed that there are some habitats that were ideally suited for one microbe or another, and yet these microbes were not present.
He offered the following explanation: “There thus are rare and less rare microbes. Perhaps there are very rare microbes, i.e., microbes whose possibility of dispersion is limited for whatever reason.”
Useful models are never “true” in the usual sense of the word. Models like the Baas Becking hypothesis divide the world into distinct intellectual habitats; one in which the model holds, and one in which it doesn’t. At the shore between the two habitats, there is an intellectual littoral zone; a place where the model gives way, and something else rises up. As any naturalist knows, most of the action happens at interfaces; land and sea, sea and air, sea and mud, forest and prairie. The principle applies just as well to the landscape of ideas. The limits of a model, especially one as sweeping as Baas Becking’s, provides a lot of cozy little tidal ponds for graduate students to scuttle around in.
By the way, guess where Lourens Baas Becking first developed his hypothesis? He was here in California, studying the halopiles of the local salt lakes. In fact, the very ones I will be studying.












That was very fun to read Russell. I think you have a compelling project and it should be exciting for all of us to see your results. In terms of your writing, I would suggest that you state your hypothesis more directly (don't bother with “I think that”) and if you can be more specific, do so. I.e., what do you mean by “major role”?
Good luck!
-Cameron Thrash
LikeLike
Cameron —
Thanks for the support! It's always nice to know I'm not the only one who thinks this is interesting.
To answer your question, I suppose my hypothesis would be better stated that dispersal plays ANY role in community formation. As things stand right now, I think everyone believes that it does. As far as I know, this hasn't been rigorously established for wild communities except in very special cases, like disease.
While I'm pretty confident that I will see finite dispersal rates, I have no idea what sort of process they will point to. The next step — tracking down the vectors of dispersal — is the truly exciting bit, but without the rates I can only speculate. You can guess how my committee would feel about that. 🙂
Russell
LikeLike
Cool. It will be neat to see what you come up with. Yes, finding vectors for dispersal may be a bit beyond the scope of one dissertation!
Cameron
LikeLike
I think the most important part is the first section. If you don't get samples that are distant enough dispersal-wise, and in sufficient numbers, you will be wasting your time on this project. (Which makes me ask the question as to whether or not you have thought of alternative angles on the data that might give you something publishable after all your hard work.)
So I would question if your concentration on easily accessible locations within driving distance is appropriate, given what you are looking for.
At the least, you should be thinking about how to develop ideas for dispersal distance between sites as opposed to geographical distance.
To do that, you could do some thinking about dispersal mechanisms. Wind and rain are big ones. Clouds are full of microbes, and microbes are seeds for condensation. Birds are another, as are the feet of animals. Insects are also covered with microbes. Even the lowly aphid can migrate thousands of miles.
So you could take your proposed sites and look at them from meteorological distance, wind patterns, bird populations and migrations, insect populations. There may be dispersal maps for various bird species, for insects,etc.
But also, thinking about those listed dispersal mechanisms, there are quite good reasons to believe that Baas Becking's theory is, for practical purposes, substantially correct.
That would lead me to think that what you are most likely to find is not data overturning his theory, but rather, data showing something about how dispersal works, and perhaps correlating that with various mechanisms like weather patterns, etc. Weather distance is a tough one though because we get dust from the Sahara landing in the Southeast of North America sometimes. You could take a look at that literature.
I would back up the research idea a bit and try to make it more inclusive. Perhaps you could also be attempting to look at dispersal mechanisms, and develop some evidence for how Becking's theory works?
And it would seem to me that you should be developing multiple data groups. For instance, it would seem to me to make more sense to collect samples from several hot spring areas that are very distance. For instance, one set from Kamchatka, another set from Yellowstone, a set from Mammoth, Beppu Japan, Reykholtsdalur, etc.
If you did that, you could at the worst back up to finding something interesting worth talking about after all your sequencing was done. Showing the tree of life for each of those springs and their relative relatedness should yield something.
If you could wangle a sample of the water from lake Vostok in Antarctica, instead of examining thermophiles, you could examine freshwater psychrophiles instead, using a similar strategy.
But, you did have a good point about conditions needing to be identical. So maybe you might want to look at something a bit different. You said that there are places where microbes are not found that could be expected. Maybe concentrate on those environments and sequence them. You should find some sort of relationship.
I'm also wondering if, in this area, it is necessarily the most productive to overlay the artifice of requiring an hypothesis that must be proved or disproved to be asked in front of your fishing expedition. I know it is de rigeur for the most part in biological sciences these days, but it wasn't always that way.
It used to be that bioscience projects could be pursued much as astronomy still is. Just go look and see what you turn up. Some very useful papers have come from that. Jonathan may want to duct tape me for saying that. But considering what metagenomics is, I see it as more observational science than hypothesis driven.
Oh – select your committee well – to the extent you can. 🙂
LikeLike