 Good to see here that Simon Chan, from UC Davis, knows what is the best outfit to wear to present his work to Bill Gates.
Good to see here that Simon Chan, from UC Davis, knows what is the best outfit to wear to present his work to Bill Gates. Thanks to Simon for sending me the photo and to the Bill and Melinda Gates Foundation for approving it’sits use here.
 Good to see here that Simon Chan, from UC Davis, knows what is the best outfit to wear to present his work to Bill Gates.
Good to see here that Simon Chan, from UC Davis, knows what is the best outfit to wear to present his work to Bill Gates. Thanks to Simon for sending me the photo and to the Bill and Melinda Gates Foundation for approving it’sits use here.
There has been a lot of hand wringing over whether we should and if we should how should we link discussions about scientific papers with the papers themselves. For example, if someone writes a news story about a paper in BMC Genomics – should the online version of the paper show links to the news story? I think so, as so many others. If someone writes a blog post discussing the paper, should there be a tracked link on the journal site? Again, I think so. I think this is even more important in social media discussions of papers, which I find fascinating much of the time. Very few people go to journal sites and post comments and open up discussions of papers. But lots of people post comments to twitter, facebook, and other social media sites. So why not bring those posts into the fold?
Now there are lots of efforts out there to collect comments about papers. Faculty of 1000. The Third Reviewer. Research blogging. And much more. For other discussions of the issue see:
My simple suggestion:
If you see ANY online discussion of a paper – a news story – a blog – even some smaller thread somewhere. Find the journal article online and use the comments function is the journal has one to post a comment saying “There was a news story discussing this paper in the New York Times. See ….” Or something like that. And presto, people who go see the paper online will also have potential to find the link you post.
I have been doing this for a while. It is relatively easy for PLoS Papers. For example for my paper on “Stalking the Fourth Domain in Metagenomic Data” I posted all sorts of links using the PLoS One comment function. I posted links to my blog. I posted links to positive news stories. I posted links to critiques. Anything I could find.
And this worked out pretty well.
I then started posted links for other papers, even pretty old ones (I just posted a few for my PLoS Biology paper in 2006 on the Tetrahymena genome). I have now done this for many PLoS papers as well as my recent Nature paper on a “Phylogeny driven genomic encyclopedia of bacteria and archaea“. Now, mind you, this works best when the papers are open access or at least freely available, so that people can read the paper as well as the discussions. But you could do this for any paper in principle if the journal has a commenting function.
Now – I am not alone in starting to do this. PLoS One has even launched this as a formal “media tracking” project: see PLoS ONE’s Media Tracking Project | EveryONE. Not sure how well their system will work, but any aggregation is good. Of course, in the long run, systems that aggregate automatically using trackbacks or DOIs or other tools will be best (e.g., some journals link to Research Blogging posts but not all do), but those do not always work perfectly and some journals do not seem to like the automated approaches. So please – link and comment away. Become part of the aggregation solution. I know this is not all we need to do and this is a relatively minor thing – but if we get everyone engaged in doing it, I believe there will be a catalytic effect whereby people will then understand why this might be useful to do broadly …
Here is the abstract:
“Despite the power of massively parallel sequencing platforms, a drawback is the short length of the sequence reads produced. We demonstrate that short reads can be locally assembled into longer contigs using paired-end sequencing of restriction-site associatedDNA (RAD-PE) fragments. We use this RAD-PE contig approach to identify single nucleotide polymorphisms (SNPs) and determine haplotype structure in threespine stickleback and to sequence E. coli and stickleback genomic DNA with overlapping contigs of several hundred nucleotides. We also demonstrate that adding a circularization step allows the local assembly of contigs up to 5 kilobases (kb) in length. The ease of assembly and accuracy of the individual contigs produced from each RAD site sequence suggests RAD-PE sequencing is a useful way to convert genome-wide short reads into individually-assembled sequences hundreds or thousands of nucleotides long.”
Note as they note in the paper “Competing interests: E.A.J. has patents filed on the RAD marker, and partial interest in a company commercializing the system. This does not alter the authors’ adherence to all the PLoS ONE policies on sharing data and material” This seems like it would have potential in metagenomic applications. I note, we are working on a similar approach – and kind of got scooped here in a way. Hope their patent does not limit what we can do.
![]()
And the bad new omics words keep streaming in. Today’s winner of the “Worst New Omics Word Award” is going to Carey Lambert, Chien-Yi Chang, Michael J. Capeness and R. Elizabeth Sockett from Nottingham for their use/ invention of “Predatosome”. They use this term in the title of their new PLoS One paper: The First Bite— Profiling the Predatosome in the Bacterial Pathogen Bdellovibrio. Here is the very long sentence where the define it:
The gene products required for the initial invasive predatory processes have not been extensively studied but the genome sequencing of B. bacteriovorus HD100 [1] revealed a genome of 3.85Mb, including a core genome similar to that of non-predatory bacteria and some 40% of the genome comprising a potential predicted “predatosome” of genes, encoding both hydrolytic products that may be employed in prey degradation, and genes that may be required specifically for host predation and thus are not conserved across the Proteobacteria.
The paper overall is quite nice on first read. They used microarray studies to characterize gene expression patterns in different phases of the life cycle (see Figure above for the life cycle outline). They backed up this work by quantitative PCR studies and regular RT PCR. And based upon their analysis they found some genes that are “Up-Regulated in Predatory, but Not HI” phase (HI stands for host-independent). And here is where they really tell us what they mean by predatosome:
This category of 240 genes are very interesting as they potentially exclude those genes simply involved with release from attack-phase into growth, namely they should be part of the “predatosome” of predatorily specific genes.
It seems to me this terminology is completely unnecessary. All they need to do is say they are studying the genes related to the predatory phase. To assign these genes to the “predatosome” is a bit much. They continue in the paper to report some really interesting stuff. For example, they also examine another predatory bacterial species, and look at whether there are genes conserved in the process between species. They made some really nice figures by the way about the different phases of hte life cycle in this organism and which genes are expressed:
Lambert, C., Chang, C., Capeness, M., & Sockett, R. (2010). The First Bite— Profiling the Predatosome in the Bacterial Pathogen Bdellovibrio PLoS ONE, 5 (1) DOI: 10.1371/journal.pone.0008599
Friendfeed comments below:
http://friendfeed.com/treeoflife/9262afad/cool-paper-winner-of-worst-new-omics-word-award?embed=1
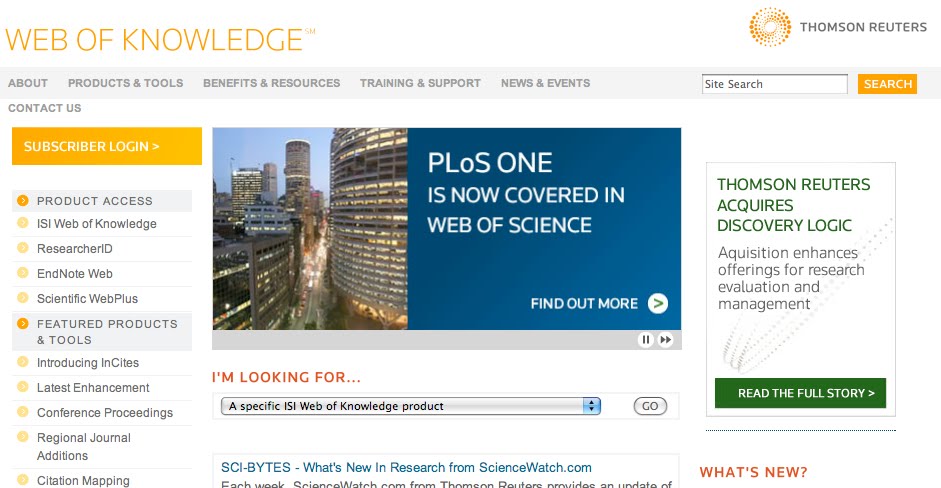 Well, just a mini post here. In case you did not know – PLoS One is now being indexed by ISI (see their announcement: PLoS ONE and see the PLOS blog post here
Well, just a mini post here. In case you did not know – PLoS One is now being indexed by ISI (see their announcement: PLoS ONE and see the PLOS blog post here
and see Erik Svensson’s blog post for an interesting take) and will get an impact factor and be in their Citation Index and all such things. Now mind you, I think “Impact Factor” is a silly thing overall in that we should evaluate papers not journals per se.
So why am I writing this – because I find it pretty funny that despite being slow to recognize PLoS One ISI is now promoting the fact that they are indexing PLoS One on their home page. See the screen capture above.
http://friendfeed.com/treeoflife/fa0c4b0b/isi-late-to-index-plos-one-but-now-marketing-that?embed=1
UPDATE – READ COMMENTS – LEAD AUTHOR HAS GOTTEN PRESS RELEASE CHANGED
A new paper just showed up on PLoS One and it has some serious potential to be important The paper (PLoS ONE: The Effects of Circumcision on the Penis Microbiome) reports on analyses that show differences in the microbiota (which they call the microbiome – basically what bacterial species were present) in men before and after circumcision. And they found some significant differences. It is a nice study of a relatively poorly examined subject – the bacteria found on the penis w/ and w/o circumcision. This is a particularly important topic in light of other studies that have shown that circumcision may provide some protection against HIV infection.
In summary here is what they did – take samples from men before and after circumcision. Isolate DNA. Run PCR amplification reactions to amplify variable regions of rRNA genes from these samples. Then conduct 454 sequencing of these amplified products. And then analyze the sequences to look at the types and #s of different kinds of bacteria.
What they found is basically summarized in their last paragraph
“This study is the first molecular assessment of the bacterial diversity in the male genital mucosa. The observed decrease in anaerobic bacteria after circumcision may be related to the elimination of anoxic microenvironments under the foreskin. Detection of these anaerobic genera in other human infectious and inflammatory pathologies suggests that they may mediate genital mucosal inflammation or co-infections in the uncircumcised state. Hence, the decrease in these anaerobic bacteria after circumcision may complement the loss of the foreskin inner mucosa to reduce the number of activated Langerhans cells near the genital mucosal surface and possibly the risk of HIV acquisition in circumcised men.”
And this all sounds interesting and the work seems solid. I note that some friends / colleagues of mine were involved in this including Jacques Ravel who used to be at TIGR and now is at U MD and Paul Kiem who is associated with TGen in Arizona. For anyone interested in HIV, the human microbiome, circumcision, etc, it is probably worth looking at.
However, the press release I just saw from TGen really ticked me off. The title alone did me in “Study suggests why circumcised men are less likely to become infected with HIV”. Sure the study did suggest a possible explanation for why circumcised men are less likely to get HIV infections – the paper was justifiably VERY cautious about this inference. They basically state that there are some correlations worth following up.
The press release goes on to say “The study … could lead to new non-surgical HIV preventative strategies for the estimated 70 percent of men worldwide (more than 2 billion) who, because of religious or cultural beliefs, or logistic or financial barriers, are not likely to become circumcised.” Well sure, I guess you could say that. I think they are iplying you could change the microbiome somehow and therefore protect from HIV but that implies (1) that there really is a causal relationship between the microbial differences in HIV protection and (2) that one could change the microbiome easily, which is a big big stretch given how little we know right now.
Anyway – the science seems fine and not over-reaching. But the press release is annoying and misleading. Shocking I know. But this one got to me.
UPDATE – SEE COMMENTS HERE AND IN FRIENDFEED. LEAD AUTHOR GOT PRESS RELEASE CHANGED.
![]()
Price, L., Liu, C., Johnson, K., Aziz, M., Lau, M., Bowers, J., Ravel, J., Keim, P., Serwadda, D., Wawer, M., & Gray, R. (2010). The Effects of Circumcision on the Penis Microbiome PLoS ONE, 5 (1) DOI: 10.1371/journal.pone.0008422
My lab has a new paper that just came out on the sequencing and analysis of the genome of a pretty cool (or hot actually) bacterium, Thermomicrobium roseum, which was isolated from a Toadstool Spring, an alkaline siliceous hotspring in Yellowstone National Park. This paper is from a grant we had when I was at TIGR as part of the “Assembling the Tree of Life” program at NSF. Our grant was focused on generating genome sequences from phyla of bacteria for which no genomes were available.
So I am offering up my paper as a case study. If you comment and ask questions or make critiques, I will try to respond. And if you think something in our paper is wrong or weird, please say so. If you think something in our paper is supported by other work we do not cite, please say this too. If you have anything useful to say, please make comments.
How do you do this?

Seen recently at the California Railroad Museum. I guess the people from Nature were right – PLoS One is leaving its mark everywhere.
Now, mind, you I like Nature as a publishing unit. They publish some very fine journals. Now, most of them are not Open Access, so I choose not to publish there if I can avoid it. But I still like them. And many of the editors and reporters there are excellent – smart, creative, insightful and such. But Nature the publisher can also be completely inane when it comes to writing about Open Access and PLoS. In a new article by Declan Butler, Nature takes another crack at the PLoS “publishing model”
![]()
Well, I have truly entered the modern world. My first PLoS One paper has just come out. It is entitled “An Automated Phylogenetic Tree-Based Small Subunit rRNA Taxonomy and Alignment Pipeline (STAP)” and well, it describes automated software for analyzing rRNA sequences that are generated as part of microbial diversity studies. The main goal behind this was to keep up with the massive amounts of rRNA sequences we and others could generate in the lab and to develop a tool that would remove the need for “manual” work in analyzing rRNAs.
We first developed this pipeline/software in conjunction with analyzing the rRNA sequences that were part of the Sargasso Sea metagenome and results from the word was in the Venter et al. Sargasso paper. We then used the pipeline and continued to refine it as part of a variety of studies including a paper by Kevin Penn et al on coral associated microbes. Kevin was working as a technician for me and Naomi and is now a PhD student at Scripps Institute of Oceanography. We also had some input from various scientists we were working with on rRNA analyses, especially Jen Hughes Martiny
We made a series of further refinements and worked with people like Saul Kravitz from the Venter Institute and the CAMERA metagenomics database to make sure that the software could be run outside of my lab. And then we finally got around to writing up a paper …. and now it is out.
You can download the software here. The basics of the software are summarized below: (see flow chart too).
From the above path, we end up with an alignment, which is useful for things such as counting number of species in a sample as well as a tree which is useful for determining what types of organisms are in the sample.
I should note this work was supported primarily by a National Science Foundation grant to me and Naomi Ward as part of their “Assembling the Tree of Life” Program (Grant No. 0228651). Some final work on the project was funded by the Gordon and Betty Moore Foundation through grant #1660 to Jonathan Eisen and the CAMERA grant to UCSD.
Wu, D., Hartman, A., Ward, N., & Eisen, J. (2008). An Automated Phylogenetic Tree-Based Small Subunit rRNA Taxonomy and Alignment Pipeline (STAP) PLoS ONE, 3 (7) DOI: 10.1371/journal.pone.0002566






