
Recently I received and email from Rachid Ounit pointing me to a new open access paper he had on a metagenomics analysis tool called CLARK. I asked him if he would be willing to write a guest post about it and, well, he did. Here is it:
CLARK: Accurate metagenomic analysis of a million reads in 20 seconds or less…
At the University of California, Riverside, we have developed a new lightweight algorithm to classify accurately metagenomic samples while minimizing computational resources better than any other classifiers (e.g., Kraken). While CLARK and Kraken have comparable accuracy, CLARK is significantly faster (cf. Fig. a) and uses less RAM and disk space (cf. Fig. b-c). In default mode and single-threaded, CLARK’s classification speed is higher than 3 million short reads per minute (cf. Fig. a), and it also scales better in multithreading (cf. Fig. d). Like Kraken, CLARK uses k-mers (short DNA words of length k) to solve the classification problem. However, while Kraken and other k-mers based classifiers consider the whole taxonomy tree and must resolve k-mers that match genomes from different taxa (by using the concept of “lowest common ancestor” from MEGAN), CLARK rather considers taxa defined for a unique taxonomy rank (e.g. species/genus), and, during the preprocessing, discards any k-mers that can be found in any pair of taxon. In other words, CLARK exploits specificities of each taxon (against all others) to populate its light and efficient data structure. It uses a customized dictionary of k-mers, in which each k-mer is associated to at most one taxon and results in fast k-mer queries. Then, the read is assigned to the taxon that has the highest amount of k-mers matches with it. Since these matches are discriminative, CLARK assignments are highly accurate. We also show that the choice of the value of k is critical for the optimal performance, and long k-mers (e.g., 31-mers) are not necessarily the best choice to perform accurate identification. For example, high confidence assignments using 20-mers from real metagenomes show strong consistency with several published and independent results.
Finally, CLARK can be used for detecting contamination in draft reference genome or, in genomics, chimera in sequenced BACs. We are currently investigating new techniques for improving the sensitivity and the speed of the tool, and we plan to release a new version later this year. We are also extending the tool for comparative genomics/metagenomics purposes. A “RAM-light” version of CLARK for your 4 GB RAM laptop is also available. CLARK is user-friendly (i.e., easy to use, it does not require strong background in programming/bioinformatics) and self-contained (i.e., does not need depend on any external software tool). The latest version of CLARK (v1.1.2) contains several features to analyze your results and is freely available under the GNU GPL license (for more details, please visit CLARK’s webpage). Experimental results and algorithm details can be found in the BMC genomics manuscript.
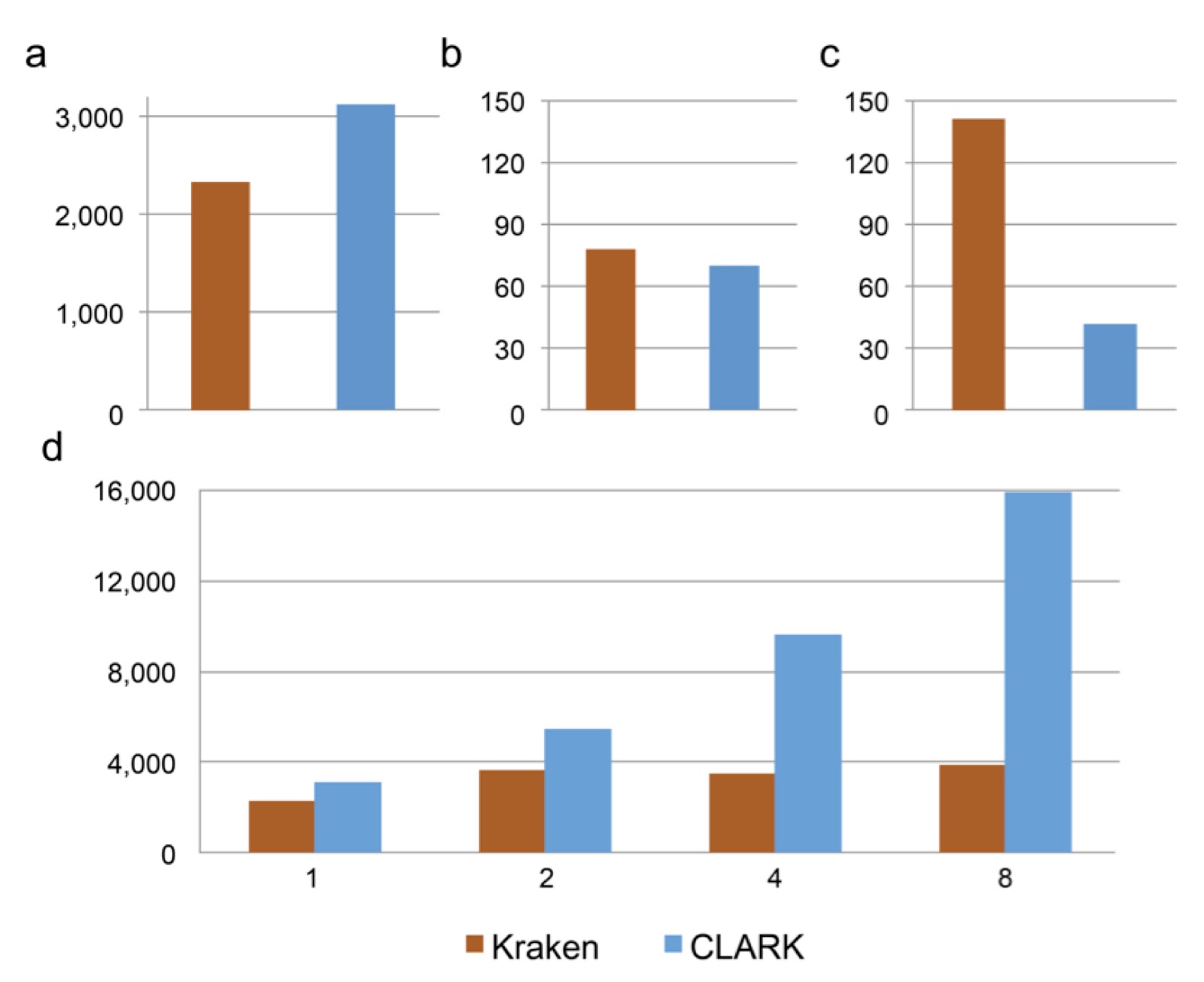 |
| Performance of Kraken (v0.10.4-beta) and CLARK (v1.0) for the classification of a metagenome sample of 10,000 reads (average reads length 92bp). a) The classification speed (in 103 reads per minute) in default mode. b) RAM usage (in GB) for the classification. c) Disk space (in GB) required for the database (bacterial genomes from NCBI/RefSeq). d) Classification speed (in 10^3 reads per minute) using 1, 2, 4 and 8 threads. |


Just FYI – The links to the website and the manuscript are broken.
LikeLike
thanks – should be fixed now
LikeLike
Looks really promising! But 30Gb disk space for just 10 thousand reads (even more for Kraken), is that really true?
LikeLike
The question is seven months old now, but I figured I'd answer it. That disk space is for the database that the reads are compared against (bacterial genomes from NCBI/RefSeq per the figure).
LikeLike