
I am very excited about today’s post. It is the first in what I hope will be many – posts from authors of interesting papers describing the “Story behind the paper“. I write extensive detailed posts about my papers and also have tried to interview others about their papers if they are relevant to this blog. But Matthew Hahn approached me recently about the possibility of him writing up some details on his recent paper on the functions of orthologs vs. paralogs. So I said “sure” and set up a guest account for him to write up his comments and details of the paper.
For those of you who do not know, Matt is on the faculty at U. Indiana. He was a post doc at UC Davis so I have a particular bias in favor of him. But his recent paper has generated some controversy (I posted some links about it here). So it is great to get some more detail from him. In addition, I note, I am also using this approach to try and teach people how easy it is to write a blog post by getting them guest accounts on Blogger and letting them write up something with links, pictures, etc. So hopefully we can get more scientists blogging too.
Anyway – without any further ado – here is Matt’s post:
———————————————————————–
Following Jonathan’s excellent
example of how explaining the history of a project helps to illuminate how the process of science actually happens, I thought I’d start by giving a bit of history behind our study, and the paper that we recently published in PLoS Computational Biology (
http://www.ploscompbiol.org/article/info:doi/10.1371/journal.pcbi.1002073). And then I’ll address the critics…
How this all got started
It all started a bit more than three years ago, in the summer of 2008. Pedja (as Predrag Radivojac is known to friends) was giving a talk to a group of us on protein function prediction that he also presented as a tutorial at the Automated Function Prediction SIG at ISMB 2008. Pedja and I had already collaborated on a small
project involving the evolution of phosphoryation sites, but I really had no idea about his work on function prediction, and little idea in general about how function prediction was done. Reviewing different ways to accomplish transfer-by-similarity, he eventually got around to evolutionary (phylogenomic) approaches. Here is what I remember of this specific exchange during his talk:
Pedja: …and of course these methods only use orthologs for prediction, because orthologs have more similar functions than do paralogs.
me (from audience): Who says?
Pedja: Umm, you say. I mean, the evolutionary biologists say.
me: No, we don’t. I don’t know of any data that says any such thing.
Pedja: Whatever, Matt. We’ll talk about this later.
Well, we did talk about it later, and it turned out that although this claim is made in tons of papers, there is basically no data to support it. In the best cases a real example of one gene family will be cited, but there are very few of these. In the worst cases, the authors will just cite some random paper about gene duplicates (or Fitch’s original
paper defining orthologs and paralogs). Of course I agree that patterns of
sequence evolution might lead you to conclude this relationship was true, but there was no experimental data.In fact, as we say in our paper, rarely did anyone recognize that this claim needed to be tested, or even that it was a claim that could be tested. At the time Eugene Koonin was the only person to say this: “The validity of the conjecture on functional equivalency of orthologs is crucial for reliable annotation of newly sequenced genomes and, more generally, for the progress of functional genomics. The huge majority of genes in the sequenced genomes will never be studied experimentally, so for most genomes transfer of functional information between orthologs is the only means of detailed functional characterization” (
http://www.ncbi.nlm.nih.gov/pubmed/16285863). I really liked the way that Eugene had said this, and started to refer to the idea that orthologs were more functionally similar than paralogs as the “ortholog conjecture.” So to be clear: I completely made up this phrase, but used the most evocative word from the Koonin paper.
Luckily for Pedja and me we had just gotten a small internal grant to work on genome annotation and we had an incoming master’s student (Nathan Nehrt) who was willing to work on a project intending to test the ortholog conjecture.
Interlude: the crappy state of things in the study of the evolution of function
In order to test anything about how function evolves between orthologs and paralogs—or between any genes—one of course needs some kind of data on gene function in multiple species. And this turns out to be a big problem.
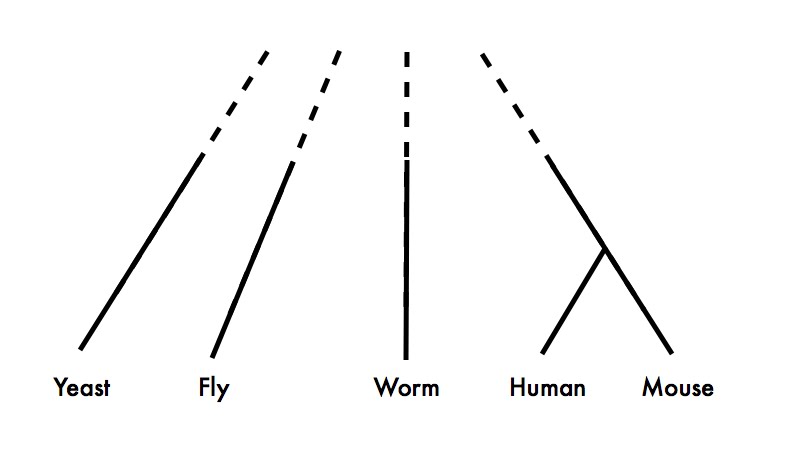
Because, as Koonin says in the earlier quote, the vast majority of experimental data comes from a very few species, and these species are not exactly closely related. Here is an approximate phylogeny of the major eukaryotic model organisms:
It’s obvious from this figure that if you need both 1) lots of functional data from two species, and 2) a pretty good idea of exactly what the homologous relationships are between the genes you’re studying, you’re going to have to study human and mouse.
This is actually a pretty bleak picture for people who study molecular evolution (as I do). While we have tons and tons of sequence data both within and between species, and a very good idea about how these sequences evolve, and fancy models with which to analyze these sequences…we know next to diddly-squat about general patterns relating these sequence differences to functional differences. There are lots of interesting things to be gleaned from studies of sequence evolution, but it really would be nice to know something about the relationship between sequence and function.
What we found
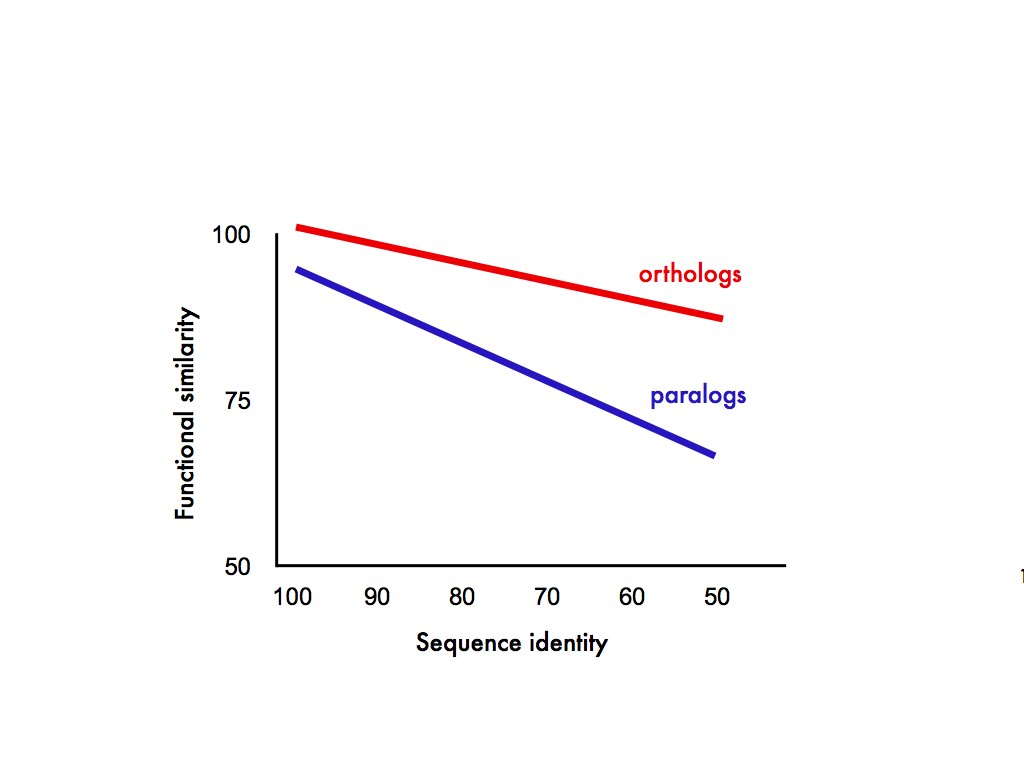
What exactly does the ortholog conjecture predict? In my mind, at least, it predicts something like this:
In this completely fictitious graph the relationship between protein function and sequence similarity is a declining one, only it declines faster for paralogs than it does for orthologs. Also, just possibly, gene duplicates start out with slightly diverged function the minute they appear. Anyway, those were our predictions.
But here is what we found (Figure 1 in Nehrt et al. 2011):
(Panel A uses the Biological Process ontology and panel B uses the Molecular Function ontology.)
There are really two different, equally surprising results here. First, there is no relationship between sequence divergence and functional divergence for orthologs (among 2,579 one-to-one orthologs between human and mouse). Absolutely none—it’s a straight horizontal line. Second, there is a relationship for paralogs (among 21,771 comparisons), exactly as we predicted there would be. So according to our results, paralogs actually have more conserved function than do orthologs. Our interpretation of the data was that the most important determinant of function was the organismal context in which a gene/protein found itself: given the same amount of sequence divergence, two proteins in the same organism would be more functionally similar. For orthologs, this means that the sequence divergence of our target gene was not the most important thing, but rather the sum total of divergence in all of the genes that contribute to its cellular context. Which is why all the orthologs have on average similar functional divergence—they are all exactly the same age and hence have approximately the same levels of divergence in these interactors (in this case sequence divergence for paralogs is a much better indicator of their splitting time).
Without going through every result in the paper and our interpretation of every result, suffice it to say that after about a year-and-a-half of working on this (around February 2010), we were satisfied that we had something we were willing to submit. I even seem to remember showing the above figure to Jonathan on a visit to UC-Davis! So we did submit the paper, first to PNAS and then, after rejection, to PLoS Computational Biology, where it was rejected again.
The content of the reviews was approximately the same at both journals. Basically, people were not convinced of our results mostly because the functional relationships were all based on data in the Gene Ontology database. To be specific, the functional data we used came from experiments conducted in 12,204 different papers. We didn’t use any predicted functions, only functions assigned using experimental data. And we did A LOT of work to try to eliminate problems that might have affected our results, including repeating the main analysis using only GO terms common to both the human and mouse datasets. But there can still be bias hidden within these functional assignments because someone always has to interpret the experiment—to say that a yeast two-hybrid experiment means that a gene has function X. And because of these biases, people weren’t buying it.
To get a measure of functional similarity that did not depend on the interpretation of any experiments, we decided to repeat the entire analysis using microarray data, using the correlation in expression levels across 25 tissues as the measure of functional similarity. By this time Nathan was graduating and moving on to Maricel Kann’s lab as a research programmer, so we recruited one of Pedja’s Ph.D. students, Wyatt Clark, to pick up where Nathan had left off. (Wyatt had actually been a student in my undergraduate Evolution course a few years earlier, so we figured he knew something…) After repeating all of the GO-based analyses himself—always better to double-check, right?—Wyatt got all of the microarray data in order and produced this figure (Figure 4 in Nehrt et al. 2011):

So a year after we first submitted a paper, we submitted a new version to PLoS CB including the array analysis, and this was enough to convince the reviewers that our results were not merely due to some strange bias in GO.
The fallout, and some responses
First, let me say that I had some idea that this would be a controversial-ish paper, and that we’d get at least some blowback. For about the first 20 versions of the manuscript (including some submitted versions) I put the words “ortholog conjecture” in quotes in the title, never an endearing move. (Pedja finally convinced me to take them out of the latest submissions.) But I also thought people would be happier that an untested assumption had finally been tested—and we have definitely gotten some positive feedback along these lines, including several groups that told us they have data that support our findings. By coincidence my lab had another paper come out the same week as this one (
http://www.ncbi.nlm.nih.gov/pubmed/21636278), and I mistakenly thought it would generate much more attention. I still think the biological importance of the results in that one are much greater than the ortholog conjecture results, but either because we didn’t publish in an open-access journal (Jonathan is always right) or simply because the function-prediction community is more active on the interweb tubes, there have been a surprising number of critical responses (partially collected here:
http://phylogenomics.blogspot.com/2011/09/some-links-on-ortholog-conjecture-paper.html). So here are some responses to general critiques.
The ortholog conjecture says only that orthologs are similar.
Okay, this one is a bit unfair, as only one person has said this. The real problem here is that Michael Galperin seems to have deeply misunderstood what we mean by the ortholog conjecture.
According to him the ortholog conjecture is “the assumption that orthologs (genes with a common origin that were vertically inherited from the same gene in the last common ancestor of the host organisms) typically retain the same function or have closely related ones.” Umm, no. In fact, if you really think this is what the ortholog conjecture says, then our results support it—human and mouse orthologs do typically have closely related functions. But we are explicitly testing for a difference between orthologs and paralogs, not whether or not orthologs retain any functions. At no point did we say (or even hint) that orthologs should not be used for functional prediction. The whole point of our analysis and conclusions is that we should stop ignoring paralogs, which would give us a ton more data to use for the prediction of functions.
The assignments of orthology and paralogy are incorrect.
This is an easy one: we did in fact get the definitions of in- and out-paralogs correct (and laid them out in Figure S1). According to Sonnhammer and Koonin: “Our definition of ‘outparalogs’ is: paralogs in the given lineage that evolved by gene duplications that happened before the radiation (speciation) event” (
http://www.ncbi.nlm.nih.gov/pubmed/12446146). For the purposes of our study, this means that outparalogs are defined as any paralogs that diverged before the speciation event between human and mouse and inparalogs diverged after this speciation event. Outparalogs do not indicate only paralogs in two different species, though by necessity in our dataset inparalogs are only found in the same species (all in human or all in mouse). Therefore, with respect to our conclusion that the most important determinant of function is which genome you are found in (i.e. context), it wouldn’t matter if we had incorrect gene trees: we would never confuse two genes in the same species (either inparalogs or some of the outparalogs) with two genes in different species (all orthologs and the remaining outparalogs).
You should have inferred functions yourselves
This is a fair suggestion, and not having enough time to annotate functions for 40,000 proteins would be a pretty weak excuse for doing good science. Instead…I’ll just say that it turns out professional curators are much better at assigning functions than even the original study authors (see
http://www.ncbi.nlm.nih.gov/pubmed/20829821). Curators have a much broader view of the whole set of terms available in any ontology, and a much more consistent idea of how to apply these terms. My favorite line from the above cited article: “…because of the relatively low accuracy of the authors’ submissions, the use of authors’ annotations did not result in saving of curators’ time…”
GO is not appropriate for this analysis because it is biased.
This is the most frustrating criticism of our study, if only because it’s partly true: GO is biased. In our paper we actually detail several of these biases, including the observation that the same set of authors will give two proteins more-similar functions than will two different sets of authors. We tried very hard to attempt to control for these biases, though of course one cannot account for all of them. The most uncharitable part of this critique, however, has to be the fact that people conveniently forgot to say that our array analysis was completely distinct from the GO-based analysis (though it has its own issues), and that Burkhard Rost’s analysis of protein-protein interaction (
http://www.ploscompbiol.org/article/info:doi/10.1371/journal.pcbi.0020079) was also completely free of any bias in GO and was consistent with all of our results.
More annoying than this, you’d think from some of the critiques of GO that it was some sort of fly-by-night operation that no one should ever depend on. I mean, c’mon—there are human curators and human experimenters and of course they’re all biased so badly one could never compare functions between proteins much less between species. What were we thinking? (Only that the original GO paper has been cited >7000 times.) Funnily enough, at several points during the course of this work Pedja suggested—only half-jokingly—that we should just assume the ortholog conjecture was correct and write a paper about how GO must be wrong. Seriously, though: one would think from the excuses people came up with for the problems inherent in GO that we should simply stop using it to, you know, predict function in other species. And we were applying it to two relatively closely related mammals, one of which is explicitly a model for the other.
What next?
Our paper laid out several explicit hypotheses about the evolution of function that arose from our findings. Unfortunately, testing any of these hypotheses will require a ton more functional data, in more than one species. I know there are multiple groups working to collect these sorts of labor-intensive datasets, and Pedja and I are thinking about doing it ourselves (with collaborators, of course!). Massive datasets that reveal protein function will always be a lot harder to collect than sequence data, especially ones free from biases.
So let’s get to it…
—————————
Note – Toni Gabaldón was trying to post a detailed response but Blogger kept cutting him off with a character limit. So I have posted his response below.
I appreciate the effort by Matthew Hahnn on explaining the story behind his paper on the so-called “Ortholog conjecture” and on facing some of the criticism. This paper attracted my interest as that of many others that work on or just use orthology. For instance it was chosen by one of my postdocs for our “Journal Club” meeting. And it was discussed during our last “Quest for Orthologs” meeting in Cambridge. I think is raising a necessary discussion and therefore I think is a good paper. This does not mean that I fully agree with the interpretation and conclusions ;-). I hope to modestly contribute to this debate with the following post.
I think one of the causes that this paper has caused so much debate is that the conclusions seem to challenge common practice (inferring function from orthologs), and could be interpreted as the need of changing the strategies of genome annotation. I think, however, that one should interpret carefully these results before start annotating based on paralogous proteins. As I will discuss below one of the problems is that we need to agree in what is the conjecture to then agree in how to test it. I see three main points that can be a source of confusion: i) the issue of what is actually stated by this conjecture, ii) the issue of annotation, and iii) the issue of time
1) What is the “ortholog conjecture”?
Or in other terms, when should we expect orthologs to be more likely to share function than paralogs?. Always? Of course not. All of us would agree that two recently duplicated paralogs are likely to be more similar in function than two distant orthologs, so it is obvious that the conjecture is not simply “orthologs are more similar in function than paralogs”. In reality the expectation that orthologs are more likely to be similar in function than paralogs, as least this is how I interpret it, is directly related to the effect that duplication have on functional divergence. If gene duplication has some effect on functional divergence (even in not 100% of the cases), then, given all other things equal (divergence time, story of speciation/duplication events – except fpr the duplication defining the orthologs) one would expect orthologs to be more likely to conserve function.
I think this complexity is not well considered (by many authors, in general). Hahn refeers to the famous review of orthology by Koonin (2005) as the source for the term “ortholog conjecture”. However, In that paper this conjecture is discussed always within the context of genes accross two particular species, whether in Hahn’s paper it is taken as well to other contexts. Thus, the proper context in which to test this conjecture is only between orthologs and between-species paralogs. As we can see, Red and purple lines in Hahn paper in figure2 do not show any clear difference.
Secondly, Koonin was very cautions in his paper, stating that he was referring to “equivalent functions” and not exactly the same “function”, correctly implying that the functional contexts would be different in the two different species. This brings me to the next point.
ii) annotation
If the expectation of functional conservation of orthologs refers to a given pair of species, then it makes no sense to test that expectation between paralogs within the same species and orthologs in different species. We were interested in this issue and it took us some effort to control for this “species” influence on the comparison, if you are interested you can read our paper on divergence of expression profiles between orthologs and paralogs (http://www.ncbi.nlm.nih.gov/pubmed/21515902)
As Hahn founds, and it was anticipated by Koonin in that review, there is a huge influence of the “species context”, a big constraint of what fraction of the function is shared. Indeed I think is the dominant signal in Hahn’s paper. Why is that? One possibility is that the functional context determines the function, I agree. However, we should not discard biases in how different communities working around a model species define processes and function, also the type of experiments that are usually done. For instance experimental inference from KO mutants might be common from mouse, but I guess is not the case in humans (!!). I think this may be having a big influence and might even be the dominant signal in Hahns paper.
Finally function has many levels and I expect subfunctionalization mostly affect lower levels (i.e. more specific). Biases may also
exist in the level of annotation between species or between families of different size (contributing more or less to the orthologs/paralogs class).
Microarray data are less likely to be subject to biases (although some may exist), at least they should be expected to be free of “human interpretation biases” and so Hahn and colleaguies did well, in my opinion, of testing that dataset. It is important to note that for microarrays and for orthologs and between-species paralogs (which I think is the right frame for testing the conjecture) ortholgs are more likely to share an expression context. This is compatible to what we found in the paper mentioned above, and compatible with the orthology conjecture as stated by koonin (accross species)
iii) time
Finally, one aspect which I think is fundamental is the notion of “divergence time”. Since paralogs can emerge at different time-scales they are composed by a heterogeneous set of protein pairs. Most of comparisons of orthologs and paralogs (Hahn’s as well) use sequence divergence as a proxy of time. However this is only a poor estimate, specially when duplications (as in here) are involved (we explored this issue in the past: http://www.ncbi.nlm.nih.gov/pubmed/21075746). This means that for a given divergence time paralogs may have larger sequence divergence than orthologs at the same divergence time, or otherwise (if gene conversion is playing a role). Is the conjecture based on sequence divergence or on divergence time?, I think the initial sense of using orthology to annotate accross species is based on the notion of comparing things at the same evolutionary distance. Thus basing our conclusions on divergence times might not be the proper way of doing it.
CONCLUSIONS AND PROPOSAL FOR RE-STATEMENT
To conclude, and with the intention of going beyond this particular paper,
I would finish by saying that the key to the problem lies on how we interpret the so-called “ortholog conjecture” or how are our expectations on how function evolves. What I get from re-reading Eugene Koonin’s paper and how I am using that “assumption” in my day-to-day work is the following:
“Orthologs in two given species are more likely to share equivalent functions than paralogs between these two species”
Therefore the notion of “accross the same pair of species” is important and thus only part of the comparisons made by Hahn and colleagues could directly test this. Looking at the microarray and between-species comparisons data, the conjecture may even hold true!!
I, however, do think that the conjecture as stated above is limited and does not capture the complexity of orthology relationships. Indeed us, and many other researchers, are tuning the confidence of the orthology-based annotation based on whether the orthologs are one-to-one, one-to-many or many-to-many, even when orthologs are “super-orthologs” (with no duplication event in the lineages separating the two orthologs).
Since, the underlying assumption of the ortholog conjecture is that duplication may (not necessarily always) promote functional shifts, then many-to-many orthology relationships will tend to include orthologous pairs with different functions.
Thus I would re-state the conjecture (or expectation) as follows:
“In the absence of additional duplication events in the lineages separating them, two orthologous genes from two given species are more likely to share equivalent functions than two paralogs between these two species”
This would be a more conservative expectation, which is closer to the current use of orthology-based annotation that tends to identify one-to-one orthologs, rather than any type.
When duplications start appearing in subsequent lineages thus creating one- or many-to-many orthology relationships, the situation is less clear. Following the assumption that duplications may promote functional divergence. Then one could expand the conjecture by “the more duplications in the evolutionary history separating two genes, the lower the expectation that these two genes would share equivalent functions”.
I wrote this contribution on the fly, and surely there are ways of expressing this in more appropriate terms. In any case I hope I made clear the idea that the conjecture emerges from the notion of duplications causing functional shifts and that our expectations will be clearer if expressed on those terms. This goes on the lines of what Jonathan Eisen mentioned on considering the whole phylogenetic story to annotate genes.
Under this perspective, the real important hypothesis is that “duplications tend promote functional shifts”, I think this is based on solid grounds and has been tested intensively in the past.
Cheers,
Toni Gabaldón
http://treevolution.blogspot.com
Author: Jonathan Eisen
I am an evolutionary biologist and a Professor at U. C. Davis. (see my lab site here). My research focuses on the origin of novelty (how new processes and functions originate). To study this I focus on sequencing and analyzing genomes of organisms, especially microbes and using phylogenomic analysis
View all posts by Jonathan Eisen







{kind=link}
{kind=link}
Thanks for this post, Matt (and thanks Jonathan for inviting Matt).
I think the paper is provocative and interesting. However, I have to say, I think the microarray results *weaken* the paper, and (in that context) it's interesting to note that the microarray data were added to the paper in order to appease critical reviewers.
Why do I think the microarray results are a red herring? Because microarrays reflect transcript abundance, and control of transcription has nothing to do with the encoded protein sequence. Yet your entire investigation is about protein similarity!
Transcriptional control is driven by the promoter; so, all that the microarray results are showing is (presumably) how similar the promoters are for the various different genes (and, for orthologs, how similar the associated transcriptional machinery is in the different organisms).
Arguably, this is also what I'd expect to be the main driver of the biological process ontology associations, since (as I understand them) biological process ontology terms typically reflect highly regulated and concerted activities at the “systems” level — i.e. highly contextual activities — not individual protein activities taken in isolation.
I'm more surprised (and therefore more delighted) by your result about the molecular function ontology associations, as I probably *would* have expected protein sequence to be a bigger driver of this. But the results are not inexplicable, as I think one *would* expect that the context in which a multi-affinity enzyme is expressed could affect the observed molecular function quite a lot. This paper provides some indirect evidence for that, I think.
The bottom line, for me, is the confusion (in the field generally) between protein sequence and gene function. The gene contains more than just the protein: it includes regulatory signals too, and we know that evolution of regulation is much more significant (certainly in more “complex” organisms) than evolution of enzyme function.
In this context, I think using microarray results as a proxy for function is misleading, as the central dogma says that the protein sequence should have virtually zero direct effect on transcript expression. Protein sequence similarity may be weakly *correlated* with promoter similarity, and perhaps this is what you are measuring, but I think using *transcript* abundance as a proxy for *protein* function just muddies the issue.
LikeLike
OK – First. Thanks Matt for writing this all up.
Second – I am only going to comment very briefly here b/c alas I have pneumonia and am hacking like crazy right now.
I just want to say one thing for now. The WHOLE point of my “phylogenomic” functional prediction approach was to use evolutionary character state reconstruction as a tool in functional prediction. This approach works in the following way
1) Build a tree of genes of interest
2) Overlay experimentally determined functions onto the tree
3) Use character state reconstructions to infer functions of unknowns
This only works if function maps well onto the tree. I never really cared much about orthologs and paralogs in this. Though I did (at the insistence of reviewers in many cases) include things about orthologs. But for example, in my 1995 paper on the SNF2 family, my focus was on whether function mapped well onto the tree of the genes. I think I did not even mention orthologs or paralogs. I definitely did suggest in latter papers that one could use orthology relationships to help predict function, but that was kind of a side story and also was really dealing with the issue of ancient paralogous relationships and not recent ones.
I will comment more later about my findings on orthologs vs. paralogs in these papers, but regardless, my whole schtick has always been that one should not make general rules about protein evolution or gene evolution but should use specific information about each gene family and how function evolves in that gene family in order to predict functions of unknowns.
For example, in my very long paper on evolution of repair (Eisen and Hanawalt 1999) I wrote
“The fifth major step in phylogenomic analysis involves studies of functional evolution for individual gene families. Functional evolution was studied by overlaying information on gene functions onto the gene trees. Then parsimony reconstruction methods were used to trace changes of function over evolutionary time (this was done in much the same was as for presence and absence of genes described above). Tracing functional evolution is an important component of making functional predictions for both ancestral genes and uncharacterized genes as described (22) and phenotypic predictions for species. For example, if there have been many functional changes in the history of a particular gene or gene family, then the identification of the presence of homologs of such genes in a species is not sufficient information to predict the presence of a particular activity. Thus tracing functional changes helps prevent incorrect predictions of function. In addition, tracing functional changes can also greatly improve the chances of making correct functional predictions for ancestral genes and uncharacterized genes (22). Such functional predictions are made based on the position of the gene of interest in the gene tree relative to genes with known functions and based on identifying evolutionary events such as gene duplications that may identify groups of genes with similar functions (22).”
Each gene family is unique. General rules applied to all gene families are probably not a good idea …
LikeLike
I think it's hilarious that in Jonathan's case reviewers insisted on the inclusion of statements about orthologs and paralogs. I, of course, completely agree that an evolutionary perspective is bound to be better, though not if it's bound up by incorrect (or at least unproven) rules about which genes have more conserved function. Luckily, I think a lot of the machinery people have developed over the years for transferring character states can just as easily be used for both orthologs and paralogs (e.g. the program Sifter).
And while I agree with most of what Ian said, I don't think the transcription/protein divide is so clear. While a gene's promoter does integrate information from the cell (via transcription factors) and drive expression, it is the presence of all the different TFs that really determines expression patterns, and hence any roles that proteins can take. This obviously applies more to BP than MF, but it must contribute to the possible molecular functions a protein can take on.
LikeLike
Hi,
(Given a word limit in the comments section, I had to split my comment in two, you can read the whole piece in my blog: http://treevolution.blogspot.com/2011/09/on-orthology-conjecture.html
I appreciate the effort by Matthew Hahnn on explaining the story behind his paper on the so-called “Ortholog conjecture” and on facing some of the criticism. This paper attracted my interest as that of many others that work on or just use orthology. For instance it was chosen by one of my postdocs for our “Journal Club” meeting. And it was discussed during our last “Quest for Orthologs meeting” in cambdridge. I think is raising a necessary discussion and therefore I think is a good paper. This does not mean that I fully agree with the interpretation and conclusions ;-). I hope to modestly contribute to this debate with the following post.
I think one of the causes that this paper has caused so much debate is that the conclusions seem to challenge common practice (inferring function from orthologs), and could be interpreted as the need of changing the strategies of genome annotation. I think, however, that one should interpret carefully these results before start annotating based on paralogous proteins. As I will discuss below one of the problems is that we need to agree in what is the conjecture to then agree in how to test it. I see three main points that can be a source of confusion: i) the issue of what is actually stated by this conjecture, ii) the issue of annotation, and iii) the issue of time
1) What is the “ortholog conjecture”?
Or in other terms, when should we expect orthologs to be more likely to share function than paralogs?. Always? Of course not. All of us would agree that two recently duplicated paralogs are likely to be more similar in function than two distant orthologs, so it is obvious that the conjecture is not simply “orthologs are more similar in function than paralogs”. In reality the expectation that orthologs are more likely to be similar in function than paralogs, as least this is how I interpret it, is directly related to the effect that duplication have on functional divergence. If gene duplication has some effect on functional divergence (even in not 100% of the cases), then, given all other things equal (divergence time, story of speciation/duplication events – except fpr the duplication defining the orthologs) one would expect orthologs to be more likely to conserve function.
I think this complexity is not well considered (by many authors, in general). Hahn refeers to the famous review of orthology by Koonin (2005) as the source for the term “ortholog conjecture”. However, In that paper this conjecture is discussed always within the context of genes accross two particular species, whether in Hahn's paper it is taken as well to other contexts. Thus, the proper context in which to test this conjecture is only between orthologs and between-species paralogs. As we can see, Red and purple lines in Hahn paper in figure2 do not show any clear difference.
Secondly, Koonin was very cautions in his paper, stating that he was referring to “equivalent functions” and not exactly the same “function”, correctly implying that the functional contexts would be different in the two different species. This brings me to the next point.
(see next comment)
LikeLike
(part two)
ii) annotation
If the expectation of functional conservation of orthologs refers to a given pair of species, then it makes no sense to test that expectation between paralogs within the same species and orthologs in different species. We were interested in this issue and it took us some effort to control for this “species” influence on the comparison, if you are interested you can read our paper on divergence of expression profiles between orthologs and paralogs (http://www.ncbi.nlm.nih.gov/pubmed/21515902)
As Hahn founds, and it was anticipated by Koonin in that review, there is a huge influence of the “species context”, a big constraint of what fraction of the function is shared. Indeed I think is the dominant signal in Hahn's paper. Why is that? One possibility is that the functional context determines the function, I agree. However, we should not discard biases in how different communities working around a model species define processes and function, also the type of experiments that are usually done. For instance experimental inference from KO mutants might be common from mouse, but I guess is not the case in humans (!!). I think this may be having a big influence and might even be the dominant signal in Hahns paper.
Finally function has many levels and I expect subfunctionalization mostly affect lower levels (i.e. more specific). Biases may also
exist in the level of annotation between species or between families of different size (contributing more or less to the orthologs/paralogs class).
Microarray data are less likely to be subject to biases (although some may exist), at least they should be expected to be free of “human interpretation biases” and so Hahn and colleaguies did well, in my opinion, of testing that dataset. It is important to note that for microarrays and for orthologs and between-species paralogs (which I think is the right frame for testing the conjecture) ortholgs are more likely to share an expression context. This is compatible to what we found in the paper mentioned above, and compatible with the orthology conjecture as stated by koonin (accross species)
iii) time
Finally, one aspect which I think is fundamental is the notion of “divergence time”. Since paralogs can emerge at different time-scales they are composed by a heterogeneous set of protein pairs. Most of comparisons of orthologs and paralogs (Hahn's as well) use sequence divergence as a proxy of time. However this is only a poor estimate, specially when duplications (as in here) are involved (we explored this issue in the past: http://www.ncbi.nlm.nih.gov/pubmed/21075746). This means that for a given divergence time paralogs may have larger sequence divergence than orthologs at the same divergence time, or otherwise (if gene conversion is playing a role). Is the conjecture based on sequence divergence or on divergence time?, I think the initial sense of using orthology to annotate accross species is based on the notion of comparing things at the same evolutionary distance. Thus basing our conclusions on divergence times might not be the proper way of doing it.
LikeLike
This comment has been removed by the author.
LikeLike
ii) annotation
If the expectation of functional conservation of orthologs refers to a given pair of species, then it makes no sense to test that expectation between paralogs within the same species and orthologs in different species. We were interested in this issue and it took us some effort to control for this “species” influence on the comparison, if you are interested you can read our paper on divergence of expression profiles between orthologs and paralogs (http://www.ncbi.nlm.nih.gov/pubmed/21515902)
As Hahn founds, and it was anticipated by Koonin in that review, there is a huge influence of the “species context”, a big constraint of what fraction of the function is shared. Indeed I think is the dominant signal in Hahn's paper. Why is that? One possibility is that the functional context determines the function, I agree. However, we should not discard biases in how different communities working around a model species define processes and function, also the type of experiments that are usually done. For instance experimental inference from KO mutants might be common from mouse, but I guess is not the case in humans (!!). I think this may be having a big influence and might even be the dominant signal in Hahns paper.
Finally function has many levels and I expect subfunctionalization mostly affect lower levels (i.e. more specific). Biases may also
exist in the level of annotation between species or between families of different size (contributing more or less to the orthologs/paralogs class).
Microarray data are less likely to be subject to biases (although some may exist), at least they should be expected to be free of “human interpretation biases” and so Hahn and colleaguies did well, in my opinion, of testing that dataset. It is important to note that for microarrays and for orthologs and between-species paralogs (which I think is the right frame for testing the conjecture) ortholgs are more likely to share an expression context. This is compatible to what we found in the paper mentioned above, and compatible with the orthology conjecture as stated by koonin (accross species)
iii) time
Finally, one aspect which I think is fundamental is the notion of “divergence time”. Since paralogs can emerge at different time-scales they are composed by a heterogeneous set of protein pairs. Most of comparisons of orthologs and paralogs (Hahn's as well) use sequence divergence as a proxy of time. However this is only a poor estimate, specially when duplications (as in here) are involved (we explored this issue in the past: http://www.ncbi.nlm.nih.gov/pubmed/21075746). This means that for a given divergence time paralogs may have larger sequence divergence than orthologs at the same divergence time, or otherwise (if gene conversion is playing a role). Is the conjecture based on sequence divergence or on divergence time?, I think the initial sense of using orthology to annotate accross species is based on the notion of comparing things at the same evolutionary distance. Thus basing our conclusions on divergence times might not be the proper way of doing it.
(continues in the next message)
LikeLike
(third and last part of my message)
CONCLUSIONS AND PROPOSAL FOR RE-STATEMENT
To conclude, and with the intention of going beyond this particular paper,
I would finish by saying that the key to the problem lies on how we interpret the so-called “ortholog conjecture” or how are our expectations on how function evolves. What I get from re-reading Eugene Koonin's paper and how I am using that “assumption” in my day-to-day work is the following:
“Orthologs in two given species are more likely to share equivalent functions than paralogs between these two species”
Therefore the notion of “accross the same pair of species” is important and thus only part of the comparisons made by Hahn and colleagues could directly test this. Looking at the microarray and between-species comparisons data, the conjecture may even hold true!!
I, however, do think that the conjecture as stated above is limited and does not capture the complexity of orthology relationships. Indeed us, and many other researchers, are tuning the confidence of the orthology-based annotation based on whether the orthologs are one-to-one, one-to-many or many-to-many, even when orthologs are “super-orthologs” (with no duplication event in the lineages separating the two orthologs).
Since, the underlying assumption of the ortholog conjecture is that duplication may (not necessarily always) promote functional shifts, then many-to-many orthology relationships will tend to include orthologous pairs with different functions.
Thus I would re-state the conjecture (or expectation) as follows:
“In the absence of additional duplication events in the lineages separating them, two orthologous genes from two given species are more likely to share equivalent functions than two paralogs between these two species”
This would be a more conservative expectation, which is closer to the current use of orthology-based annotation that tends to identify one-to-one orthologs, rather than any type.
When duplications start appearing in subsequent lineages thus creating one- or many-to-many orthology relationships, the situation is less clear. Following the assumption that duplications may promote functional divergence. Then one could expand the conjecture by “the more duplications in the evolutionary history separating two genes, the lower the expectation that these two genes would share equivalent functions”.
I wrote this contribution on the fly, and surely there are ways of expressing this in more appropriate terms. In any case I hope I made clear the idea that the conjecture emerges from the notion of duplications causing functional shifts and that our expectations will be clearer if expressed on those terms. This goes on the lines of what Jonathan Eisen mentioned on considering the whole phylogenetic story to annotate genes.
Under this perspective, the real important hypothesis is that “duplications tend promote functional shifts”, I think this is based on solid grounds and has been tested intensively in the past.
Cheers,
Toni Gabaldón
http://treevolution.blogspot.com
LikeLike
(second part of my message)
ii) annotation
If the expectation of functional conservation of orthologs refers to a given pair of species, then it makes no sense to test that expectation between paralogs within the same species and orthologs in different species. We were interested in this issue and it took us some effort to control for this “species” influence on the comparison, if you are interested you can read our paper on divergence of expression profiles between orthologs and paralogs (http://www.ncbi.nlm.nih.gov/pubmed/21515902)
As Hahn founds, and it was anticipated by Koonin in that review, there is a huge influence of the “species context”, a big constraint of what fraction of the function is shared. Indeed I think is the dominant signal in Hahn's paper. Why is that? One possibility is that the functional context determines the function, I agree. However, we should not discard biases in how different communities working around a model species define processes and function, also the type of experiments that are usually done. For instance experimental inference from KO mutants might be common from mouse, but I guess is not the case in humans (!!). I think this may be having a big influence and might even be the dominant signal in Hahns paper.
Finally function has many levels and I expect subfunctionalization mostly affect lower levels (i.e. more specific). Biases may also
exist in the level of annotation between species or between families of different size (contributing more or less to the orthologs/paralogs class).
Microarray data are less likely to be subject to biases (although some may exist), at least they should be expected to be free of “human interpretation biases” and so Hahn and colleaguies did well, in my opinion, of testing that dataset. It is important to note that for microarrays and for orthologs and between-species paralogs (which I think is the right frame for testing the conjecture) ortholgs are more likely to share an expression context. This is compatible to what we found in the paper mentioned above, and compatible with the orthology conjecture as stated by koonin (accross species)
iii) time
Finally, one aspect which I think is fundamental is the notion of “divergence time”. Since paralogs can emerge at different time-scales they are composed by a heterogeneous set of protein pairs. Most of comparisons of orthologs and paralogs (Hahn's as well) use sequence divergence as a proxy of time. However this is only a poor estimate, specially when duplications (as in here) are involved (we explored this issue in the past: http://www.ncbi.nlm.nih.gov/pubmed/21075746). This means that for a given divergence time paralogs may have larger sequence divergence than orthologs at the same divergence time, or otherwise (if gene conversion is playing a role). Is the conjecture based on sequence divergence or on divergence time?, I think the initial sense of using orthology to annotate accross species is based on the notion of comparing things at the same evolutionary distance. Thus basing our conclusions on divergence times might not be the proper way of doing it.
LikeLike
This comment has been removed by the author.
LikeLike
Thanks Tony – will read in detail after a meeting — I am also going to look into the word length cut off for comments … not sure I can fix that
LikeLike
ii) annotation
If the expectation of functional conservation of orthologs refers to a given pair of species, then it makes no sense to test that expectation between paralogs within the same species and orthologs in different species. We were interested in this issue and it took us some effort to control for this “species” influence on the comparison, if you are interested you can read our paper on divergence of expression profiles between orthologs and paralogs (http://www.ncbi.nlm.nih.gov/pubmed/21515902)
As Hahn founds, and it was anticipated by Koonin in that review, there is a huge influence of the “species context”, a big constraint of what fraction of the function is shared. Indeed I think is the dominant signal in Hahn's paper. Why is that? One possibility is that the functional context determines the function, I agree. However, we should not discard biases in how different communities working around a model species define processes and function, also the type of experiments that are usually done. For instance experimental inference from KO mutants might be common from mouse, but I guess is not the case in humans (!!). I think this may be having a big influence and might even be the dominant signal in Hahns paper.
Finally function has many levels and I expect subfunctionalization mostly affect lower levels (i.e. more specific). Biases may also
exist in the level of annotation between species or between families of different size (contributing more or less to the orthologs/paralogs class).
Microarray data are less likely to be subject to biases (although some may exist), at least they should be expected to be free of “human interpretation biases” and so Hahn and colleaguies did well, in my opinion, of testing that dataset. It is important to note that for microarrays and for orthologs and between-species paralogs (which I think is the right frame for testing the conjecture) ortholgs are more likely to share an expression context. This is compatible to what we found in the paper mentioned above, and compatible with the orthology conjecture as stated by koonin (accross species)
iii) time
Finally, one aspect which I think is fundamental is the notion of “divergence time”. Since paralogs can emerge at different time-scales they are composed by a heterogeneous set of protein pairs. Most of comparisons of orthologs and paralogs (Hahn's as well) use sequence divergence as a proxy of time. However this is only a poor estimate, specially when duplications (as in here) are involved (we explored this issue in the past: http://www.ncbi.nlm.nih.gov/pubmed/21075746). This means that for a given divergence time paralogs may have larger sequence divergence than orthologs at the same divergence time, or otherwise (if gene conversion is playing a role). Is the conjecture based on sequence divergence or on divergence time?, I think the initial sense of using orthology to annotate accross species is based on the notion of comparing things at the same evolutionary distance. Thus basing our conclusions on divergence times might not be the proper way of doing it.
LikeLike
I don't know exactly why my second part of the message (I actually had to split the message in three) was not posted.
Please read the whole story here:
http://treevolution.blogspot.com/2011/09/on-orthology-conjecture.html
Jonathan if you can somehow put my comment in one piece in your blog this would be helpful.
Cheers, Toni
(this is the part that went missing)
ii) annotation
If the expectation of functional conservation of orthologs refers to a given pair of species, then it makes no sense to test that expectation between paralogs within the same species and orthologs in different species. We were interested in this issue and it took us some effort to control for this “species” influence on the comparison, if you are interested you can read our paper on divergence of expression profiles between orthologs and paralogs (http://www.ncbi.nlm.nih.gov/pubmed/21515902)
As Hahn founds, and it was anticipated by Koonin in that review, there is a huge influence of the “species context”, a big constraint of what fraction of the function is shared. Indeed I think is the dominant signal in Hahn's paper. Why is that? One possibility is that the functional context determines the function, I agree. However, we should not discard biases in how different communities working around a model species define processes and function, also the type of experiments that are usually done. For instance experimental inference from KO mutants might be common from mouse, but I guess is not the case in humans (!!). I think this may be having a big influence and might even be the dominant signal in Hahns paper.
Finally function has many levels and I expect subfunctionalization mostly affect lower levels (i.e. more specific). Biases may also
exist in the level of annotation between species or between families of different size (contributing more or less to the orthologs/paralogs class).
Microarray data are less likely to be subject to biases (although some may exist), at least they should be expected to be free of “human interpretation biases” and so Hahn and colleaguies did well, in my opinion, of testing that dataset. It is important to note that for microarrays and for orthologs and between-species paralogs (which I think is the right frame for testing the conjecture) ortholgs are more likely to share an expression context. This is compatible to what we found in the paper mentioned above, and compatible with the orthology conjecture as stated by koonin (accross species)
iii) time
Finally, one aspect which I think is fundamental is the notion of “divergence time”. Since paralogs can emerge at different time-scales they are composed by a heterogeneous set of protein pairs. Most of comparisons of orthologs and paralogs (Hahn's as well) use sequence divergence as a proxy of time. However this is only a poor estimate, specially when duplications (as in here) are involved (we explored this issue in the past: http://www.ncbi.nlm.nih.gov/pubmed/21075746). This means that for a given divergence time paralogs may have larger sequence divergence than orthologs at the same divergence time, or otherwise (if gene conversion is playing a role). Is the conjecture based on sequence divergence or on divergence time?, I think the initial sense of using orthology to annotate accross species is based on the notion of comparing things at the same evolutionary distance. Thus basing our conclusions on divergence times might not be the proper way of doing it.
LikeLike
Toni – O put your comment at the bottom of the post
LikeLike
Matt, yes – the trans-acting TFs can potentially be as important as the cis-acting promoter – but my point is that neither of these is directly determined by the protein sequence. I don't see how you can expect the protein sequence to have *any* direct predictive power about the transcription, and I think it's dubious to claim that any such correlation conveys much information. If you are seeing any correlation between protein sequence & transcription profile, it is presumably because of a mutual underlying variable in common (whether that variable is the promoter or the set of TFs in the genome). To do phylogeny on the protein sequence, then measure the transcriptional profile, then attempt to draw conclusions about “how much protein sequence tells you about function” based on these two observations (protein phylogeny & transcript abundance), is to completely ignore these hidden underlying variables (TFs and promoter). You may well observe a correlation, weak or strong, between protein phylogeny & transcription, but I don't see how you can draw a very broad conclusion about the connection between protein sequence and protein function on the basis of this. It would be like trying to describe the relationship between balls of wool and scratch-posts, without acknowledging the existence of cats.
LikeLike
Right, Ian: Time is the mutual underlying variable. I should have said that explicitly in my last comment.
LikeLike
To put it another way, I think you are introducing a NEW conjecture into the mix by doing this, or possibly two competing conjectures: (a) that protein sequences and promoter sequences share a molecular clock, (b) that changes in host TFs have a stronger effect on gene expression than changes in the promoter. This is all then tangled up with the semantics of your ontologies (BP vs MF, etc). Consequently, in my view, the transcript abundance results do not clarify the overall picture by supporting or disproving the ortholog conjecture; they merely confuse the definitions a bit more. The data and the methodology are fine in my opinion, but I think the clarity of the conclusions are possibly a bit overstated. Still a very interesting paper, and an interesting discussion
LikeLike
Just to clarify a point made by Toni: we did not take the ortholog conjecture from Koonin's 2005 paper. There are many papers that make the “orthologs are more conserved than paralogs” statement. Rather, I just liked the word “conjecture” and used it in my own way.
I think Toni is right that a simple version of the ortholog conjecture is either vacuous or obvious. Instead, here is what we said in our paper:
“Because paralogs are almost always either more- or less-related to a focal gene than an ortholog (for inparalogs or outparalogs, respectively), it is meaningless to compare the predictive power of all orthologs to all paralogs; it seems obvious that closely related orthologs will be more similar in function than distantly related paralogs, and vice versa. Instead, we focus on the predictive power of both orthologs and paralogs as a function of protein sequence divergence.”
And while Toni has certainly thought deeply about the differences in within-species and between-species paralogs, I don't think most people have. They just repeat the mantra of the ortholog conjecture. If there is a paper out there (besides Eugene's) that makes this important distinction, I have missed it.
So re-defining the ortholog conjecture to only apply to between-species comparisons is fine, as long as we make sure to tell everyone else that within-species paralogs are really well-conserved and they can stop ignoring those.
LikeLike
Oops – comments collided. Yes, time is the underlying variable… Thanks for engaging so publicly in the discussion about this, btw.
LikeLike
I agree more or less with Toni's synopsis. If people in the field are naively taking the ortholog conjecture the way that Matt suggests they are, then they aren't thinking at all about the underlying process of molecular evolution. For example, if a gene duplication mutation happened yesterday then one can expect that it would be functionally 'identical' to its parent gene assuming that the promotors/enhancers etc were all duplicated. In this case, an ortholog in an organism separated by 60 million years of evolution almost certainly MUST be more functionally divergent than the two in-paralogs. However, the excess capacity yielded by the duplicates provides the possibility of functional divergence to occur with time (subfunctionalization, neofunctionalization etc). Whether or not that happens is uncertain — there could be lots of essentially neutral duplications in genomes of individuals in populations that ultimately subsequently deleted over time or never get fixed. However, as the downward trend in funtional similarity in Matt's graph indicate when dealing with paralogs, time makes a big difference to the point where if one were to compare orthologs and paralogs at the same 'age' one does not expect to see the former as less functionally correlated than the latter.
LikeLike
I'd just like to point out that the current issue of Briefings in Bioinformatics is devoted to orthologs, including Toni's paper (congratulations Toni). http://bib.oxfordjournals.org/content/12/5.toc
LikeLike
Very interesting, thank you!
I would like to make a comment about the statement that “control of transcription has nothing to do with the encoded protein sequence”. Recent studies present a clear link between protein sequence evolutionary rate and level expression, in an anti-correlated fashion (Koonin 2011, http://tinyurl.com/3uhostg). Highly expressed genes tend to evolve slower than lowly expressed genes. Probably because of the toxicity of misfolded proteins at high level (Drummond 2008, http://tinyurl.com/6abgmyp).
LikeLike
Romain, yes there is a correlation, but it is not hypothesized to be directly causal – rather, it is due to an underlying variable in common. (In your comment, citing Eugene Koonin's paper, you posit that the level of selection is the underlying variable: highly expressed proteins are under stronger selection. In my discussion with Matt above, we referred to time as the underlying variable: similar proteins tend to have similar transcriptional machinery and promoters.) Therefore, I maintain that you cannot use transcriptional evidence to “prove” anything about the link between protein sequence and protein function. In all cases observed so far (including the paper you cite), the hypothesis has always been common causality via a third variable, rather than direct causality. I also stand by the statement I made, that the actual mechanistic *control* of transcription has nothing to do with the protein sequence (even though the observed level of transcription may be *correlated* with the conservation of the protein, for various reasons).
LikeLike
Very interesting post, thank you.
Toni, you write that
“duplications tend promote functional shifts”, I think this is based on solid grounds and has been tested intensively in the past.
Do you have citations for this? When we tried to check this a few years ago, we didn't find any good evidence, to our suprise (http://dx.doi.org/10.1016/j.tig.2009.03.004 alas not Open Access).
Given the high level of interest in this question right now, it might be a good idea to propose a dedicated symposium on this topic, for example in the upcoming SMBE or ECCB conferences.
Do others think this would be a good idea?
Marc Robinson-Rechavi
LikeLike
Hi Marc,
A terrific idea that of organizing a dedicated session on this, count on me.
>Do you have citations for this?
I just say that it has been tested many times not that the issue is completely solved. 😉 In any case, In our paper mentioned above we found an effect of duplications in terms of gene expression patterns across tissues (only one of the possible modes of functional divergence), we include some citations there of previous comparisons. There are some comparisons in terms of other types of functions or structure (see, for instance this comparison of strutural constrains betweeon ortho/paralo: http://www.ncbi.nlm.nih.gov/pubmed/19472362, or this on cellular localization: http://www.ncbi.nlm.nih.gov/pubmed/19930686). And there are many small-scale detailed examples, starting by the classical family of globins.
Duplication must not always have to promote functional shift in order for the conjecture to be true (as I expressed). Even if duplications have a small effect there would be an expected higher functional similarity for orthologs (in general terms and always expressed as a tendency that should not be true for each case) because paralogs will share the same number of events as orthologs + one duplication.
This is how I see it.
Toni
LikeLike
Just want to add something to Matt's comment.
>They just repeat the mantra of the >ortholog conjecture. If there is a >paper out there (besides Eugene's) >that makes >this important >distinction, I have >missed it.
You are right. I include myself into those that usualy do not explicitly express that distinction (never again! 😉 ). I considered enough to refer to Eugene's paper, and not reiterate everything again. I think most of the people do refer to that review, which is clear about the conjecture. But you are right in that this might not be enough and we should stress it more clearly. In general orthology-related terms and concepts suffer a lot of misuse and abuse.
Cheers, Toni
LikeLike
I was thinking more about the distinction Toni is making and was wondering what the majority of people would say about the statement:
Orthologs generally have more similar function than within-species paralogs of equal divergence.
My guess is that most people think this is true, and would not automatically give a more important role to context.
So while it's good for us to distinguish between two slightly different conjectures (orthologs vs. w/in-species paralogs and orthologs vs. btw-species paralogs), both need further examination.
And I am already planning to be in Dublin for SMBE 2012–is there still time to propose symposia?
LikeLike
Yes! The deadline for symposia is the 31st of October.
http://www.smbe2012.org/scientific-content.html
LikeLike
I think that the way Matt just put the conjecture (Orthologs generally…)is indeed the way most people see it. And which has not yet been shown to be true.
I am moving offline to try to organize a symposium proposal for SMBE. If you are interested and have not yet expressed it, please contact me directly.
LikeLike
I cannot tell what most people think. That way of putting it is definitely not how I see the conjecture, nor how it was put forward by Koonin (2005), and definitely not the rationale behind genome annotation across species.
In any case this reinforces the idea of agreeing on what are the conjectures/hypotheses. This cannot be done according to what is the most popular interpretation. Many people may still misinterpret the word orthologue to refer to genes in other species with the same function, but this doesn't change the fact that function is not what defines an orthologous pair.
Toni
LikeLike
Yes, I think we can all at least agree to permanently banish the term “functional ortholog”!
LikeLike
Very nice post and insightful discussions. I got a very naive question: how do we address domain-shuffling events in this framework?
Thanks
LikeLike
Good point!. Orthology domain shuffling complicates the issue (even conceptually, since the original definition was dealing with whole genes).
The group of Erik Sonnhammer looked at how domain architectures varied in orthologs and paralogs and found greater divergence in paralogs of a similar sequence divergence. This finding would support the conjecture.
http://www.biomedcentral.com/1471-2105/12/326
LikeLike
Thanks Toni for pointing out this paper, that I find very useful indeed. I wonder however how could we distinguish really old shuffling events (accretions) from the most recent ones. I understand mutation rates might be different, how could we then reconcile these events with the conjecture by means of the age of the genes? It may be a simple question, but I cannot figure it out.
LikeLike
In fact I was wondering to what extent this bias in GO terms come from deviations regarding domain composition, as annotations come from functional domains, it may happen that some proteins receive an annotation from similar proteins having different domain architectures (i.e: a potential pair where one member lacks a particular domain, and this is the main annotation source). Not sure if this has been corrected or taken into account while performing systematic analyses…
LikeLike
Hi Ana,
These are not simple questions. Regarding your first one I recommend a recent review by Kimmen Sjolander and colleagues, precisely on that topic:
http://bib.oxfordjournals.org/content/early/2011/06/28/bib.bbr036.full
Regarding the bias in annotation. You say annotations are based on domains, that is probably the case for “in silico” annotation. But what I understand from Matt's paper (he may clarify better than me) is that they only use annotation based on experiments and traceable author statements. If experiments are done with the whole protein then one should not expect a bias (although alternative
splicing comes to my mind as a confounding factor). In the paper of Sonnhammer they do not look at functional annotations but domain arrangements.
Toni
LikeLike
Thanks Toni:
I am not really a GO fan, but we gotta have something, and it is not easy… I understand that the common procedure for systematic GO analyses is to discard the inferred ones right? (well, this is what I do, which doesn't mean it is right either)…so, only consistent annotations should be considered.
Well, and regarding the splicing variants… you are totally right. There is a whole repertoire there frequently neglected. That's another subject. I wish they would not be neglected as usual for the sake of simplicity…
(BTW: you are a walking library ;-), just kidding).
LikeLike
I had never seen the Sonnhammer paper that Toni pointed to, and it's actually quite fascinating–inparalogs and within-species outparalogs are just as well-conserved (or more conserved) than orthologs in terms of domain architecture, at least for many millions of years after the splits. And then everything switches after that, with orthologs becoming more conserved.
This actually reminds me of a paper from Andrej Sali's lab (http://www.ncbi.nlm.nih.gov/pubmed/19472362) where the same holds true for protein structure similarity–at more recent divergences paralogs are more similar than orthologs, and then the relationships flip.
The problem is, I can't think of a good (biological) explanation for why orthologs are more similar at higher divergence levels. There is no reason to think that duplicates that have been around for many dozens of millions of years evolve any differently than “single-copy” genes.
Oh, and to answer Ana: we did indeed only use experimental codes and traceable author statements in our study (though we also did it without TAS and got the same results).
LikeLike
Hi Matthew,
thanks for your clarification.
I agree with you. I like to fantasize that maybe epigenetics has something to do with this… well, who knows.
Again, very nice and insightful post.
Thx to all.
LikeLike
Well Said. I think i m not the one who always follow up. Most people here refer to your review which is to the point about the conjecture. But the ur point of view is also right but not enough to make everything clear. People tend to misuse orthology related terms there is more useless facts common then reality among masses.
LikeLike
I leave this comment here, just to mention that this lively discussion originated the organization of a symposium in SMBE2012. You can have a look at my summary of it here, with some interesting links:
http://treevolution.blogspot.com.es/2012/06/wrap-up-of-orthology-paralogy-and.html
LikeLike
I have read with interest this story, and went to the original paper to see if it is able to change my mind (from the “ortholog conjecture”). I have a couple of criticisms. For example, when you say “Moreover, one of the three major hypotheses for the maintenance of gene duplicates (subfunctionalization) does not require any functional change”, this is not completely correct. Subfunctionalization refers to the spread of ancestral functions between gene duplicates, i.e. there is functional change in both (subfunctionalization, of course, can take place just by shifts in gene regulation, not at the protein level, what would be similar to what you said).
The other criticism is for not providing biological details. I found no example that I can think of real biological ground for your findings. That makes me doubt of the results reported. In contrast, I have in mind many real cases in which I have seen clear sequence divergence after gene duplication linked to functional changes. So, in summary, my mind refused to be changed! (not discarding the possibility, though).
One final criticism: I think your analysis would be more easier to follow if put in terms of the evolution of proteins function, and not in terms of automatic or homology-based annotation of proteins function.
And one more (last minute thought): functional divergence between paralogs is clear for deep divergence cases, right? If that is true, why more recent paralogs show a different behavior, and which would be the result in the long-term? I guess there is something contradictory there.
LikeLike
Just one more thing: an analysis based on gene ages (with respect to the gene duplication event) would be easier to follow and interpret than a comparison against sequence divergence (different rates for different genes, and not very intuitive)
LikeLike
Hi we gave a try in considering gene ages (as inferred from topology) here, in our case we did find more divergence in paralogs as compared to orthologs of a similar age:
http://bib.oxfordjournals.org/content/12/5/442.long
Evidence for short-time divergence and long-time conservation of tissue-specific expression after gene duplication
LikeLike
It is not surprising than in-paralogs are more similar than orthologs, they are just more closely related.
Regarding the long-term process, in-paralogs will be never be out-paralogs for the analyzed species… they will become out-paralogs only if the species containing them diversifies in other species… So, I do not see the problem.
Federico
LikeLike