
Just reading this paper and thought I would start a new “section” here on my blog. Journal club light. Just some notes and quick comments.
Today I am selecting this paper: Phylo SI: a new genome-wide approach for prokaryotic phylogeny. It caught my eye because, well, I am interested in genome-wide phylogeny.
So I glanced at the paper’s abstract:
The evolutionary history of all life forms is usually represented as a vertical tree-like process. In prokaryotes, however, the vertical signal is partly obscured by the massive influence of horizontal gene transfer (HGT). The HGT creates widespread discordance between evolutionary histories of different genes as genomes become mosaics of gene histories. Thus, the Tree of Life (TOL) has been questioned as an appropriate representation of the evolution of prokaryotes. Nevertheless a common hypothesis is that prokaryotic evolution is primarily tree-like, and a routine effort is made to place new isolates in their appropriate location in the TOL. Moreover, it appears desirable to exploit non–tree-like evolutionary processes for the task of microbial classification. In this work, we present a novel technique that builds on the straightforward observation that gene order conservation (‘synteny’) decreases in time as a result of gene mobility. This is particularly true in prokaryotes, mainly due to HGT. Using a ‘synteny index’ (SI) that measures the average synteny between a pair of genomes, we developed the phylogenetic reconstruction tool ‘Phylo SI’. Phylo SI offers several attractive properties such as easy bootstrapping, high sensitivity in cases where phylogenetic signal is weak and computational efficiency. Phylo SI was tested both on simulated data and on two bacterial data sets and compared with two well-established phylogenetic methods. Phylo SI is particularly efficient on short evolutionary distances where synteny footprints remain detectable, whereas the nucleotide substitution signal is too weak for reliable sequence-based phylogenetic reconstruction. The method is publicly available at http://research.haifa.ac.il/ssagi/software/PhyloSI.zip.
And something continued to catch my eye there. It was the use of “gene order conservation” as the data for the phylogenetic analysis. Hmm. I am generally skeptical of most uses of gene order for inferring phylogeny that I have seen. Why? Well, because it seems to me that gene order is less likely to be a useful character than sequences in alignments (which is the standard for inferring phylogeny). Why do I feel this way? Well, for two main reasons:
1) Sequence alignments are robust. They have been used and used and used and shown to be quite powerful and useful (even though they are not perfect). The rich literature on alignments has shown where and when and how they are useful. And where and when and how they are not. And we have powerful, tested methods to use such alignments.
2) Gene order seems less likely to be robust. I am not saying it is not useful. But the literature I have seen suggests to me that gene order is more prone to convergent evolution than sequence. And gene order is more prone to enormous variation in rates and patterns of evolution. And gene order does not actually have a lot of characters to use compared to whole genome alignment based phylogenetics.
I could go on and on. There are many other reasons I prefer sequence alignments over gene order. But I am willing to consider that gene order could be more useful than I imagine. So I read on. And the first thing I did (which is almost always the first thing I do for new phylogenetic methods papers) was I looked at their phylogenetic results. And so off to Figure 9. And the results did little to convince me that their method was better than existing alignment based methods.

I am sure people cannot see this that well. But basically, I looked through the tree and there were just too many things that are inconsistent with trees that are very supported by lots of other data.
For example
which has in one clade species that almost certainly should not group together. In particular the presence of Neisseria in this group is very strange given that all other analysis put it in the Protebacteria and the Proteobacteria are found in other parts of the tree.
And there is another clade like this
With Francisella (also considered a Proteobacteria) in a clade with things from many other Phyla.
And then there is this one.
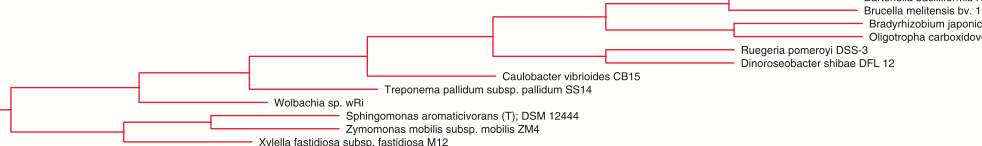
Which has gamma Proteobactera, alpha Proteobacteria, Spirochetes, and others all together in one clade.
I could go on. But this is journal club light. I just do not have time right now to dig much deeper. But on first look, I am certainly not overwhelmed with a desire to use gene order instead of sequence alignments to infer phylogenetic trees for bacteria. Again, I am not saying the method does not have its uses. It easily could be useful in many ways. But for inferring trees of all bacteria at once – does not seem to be the right thing.


We did not claim to solve all prokaryotic phylogeny with gene order. The method works very well within families and genera, where it outperforms sequence based approaches. The fact that this method works well for some phyla, and badly in others, as you pointed out, is in itself informative (indicates more rearrangements and gene insertions/deletions in their history).
LikeLike
Thanks for the comments Uri. I have looked more closely at the paper now and agree that rRNA based trees and sometimes concatenated alignment based trees can have low resolution among close relatives. But once you have a whole genome for a species one can fix those issues by using nucleotide alignments rather than amino acid alignments for concatenated trees (for example) or by adding other genes. So a better comparison to me would have been DNA alignments of protein coding genes vs. gene order. Also I do not see how gene order based analyses remove problems from HGT. Genes acquired by HGT can come in in a cluster and thus gene order would be misleading in such cases as to the species tree.
LikeLike
“the vertical signal is partly obscured by the massive influence of horizontal gene transfer (HGT)” I did not find this in the dozen or so human commensals of Neisseria at all, and I'm sure you know they're some of the most promiscuous bacteria around at least within the genus. ML, NJ, and parsimony trees of core genome were all in very good agreement and matched about all the trees I found in the literature based on 16s and even matched a lot of the old-school grouping by sugar metabolism.
LikeLike
Mary, while this may be true for some or even most commensal Neisseria, there are many that are hard to classify even using MLST data (see Figure 1 in the classic paper by Hanage, Fraser and Spratt – “Fuzzy species among recombinogenic bacteria “), which is why having a different method may be helpful.
LikeLike
While it is interesting to see whether we can detect the signal of the phylogeny-of-cells, the issue that must be faced is whether knowing it tells us what we need to know. If we are working on some protein, should be use the phylogeny-of-cells to interpret its sequences? No, not if there is a good chance that this gene had horizontal gene transfer events. Without taking that into account, the phylogeny-of-cells is much less useful in prokaryotes than in eukaryotes.
LikeLike