
//storify.com/phylogenomics/tandy-warnow-at-ucdavis-on-gene-trees-vs-species-t/embed?template=slideshow//storify.com/phylogenomics/tandy-warnow-at-ucdavis-on-gene-trees-vs-species-t.js?template=slideshow[View the story “Tandy Warnow at #UCDavis on Gene Trees vs. Species Trees” on Storify]
Tag: phylogeny

1/15 at #UCDavis: Tandy Warnow: New methods for species tree estimation in the presence of gene tree heterogeneity
Special Seminar:
Tandy Warnow
The University of Illinois at Urbana-Champaign
New methods for species tree estimation in the
presence of gene tree heterogeneity
Friday, January 15, 2016
1:30 PM
GBSF 1005
Abstract.
Estimating the Tree of Life will likely involve a two-step procedure, where in the first step trees are estimated on many genes, and then the gene trees are combined into a tree on all the taxa. However, the true gene trees may not agree with the species tree due to biological processes such as deep coalescence, gene duplication and loss, and horizontal gene transfer. Statistically consistent methods based on the multi-species coalescent model have been developed to estimate species trees in the presence of incomplete lineage sorting; however, the relative accuracy of these methods compared to the usual “concatenation” approach is a matter of substantial debate within the research community.
I will present results showing that coalescent-based estimation methods are impacted by gene tree estimation error, so that they can be less accurate than concatenation in many cases. I will also present two new methods, ASTRAL (Mirarab et al., Bioinformatics 2014) and statistical binning (Mirarab et al., Science 2014, Bayzid et al., PLOS One 2015) for estimating species trees in the presence of gene tree conflict due to ILS. Statistical binning and weighted statistical binning are used to improve gene tree estimation, while ASTRAL is a coalescent-based method that is provably statistically consistent and that can construct very accurate large species trees. Finally, I will present theoretical results investigating whether statistically consistent accurate species tree estimation is possible when gene trees have estimation error, and discuss the controversy about statistical binning (Liu and Edwards, Science 2015, Mirarab et al. Science 2015).
See Dr. Warnow’s home page for more information on her work: http://tandy.cs.illinois.edu
Host: Jonathan Eisen
Share this:
Mesquite "A modular system for evolutionary analysis" v3.0 released from Team Maddison
Just found out about this on Facebook via Rod Page: Mesquite V3.0 has been released. Mesquite is from Team Maddison (Wayne and David). I have been using their software since 1987 when I took Stephen Jay Gould’s course at Harvard and they were TAs for the course demoing an early version of MacClade. Lots of nice features and it is available in Mac, Unix/Linux, and Windows versions. They describe “What Mesquite Does” on their Wikispaces site in the following way:
Mesquite is software for evolutionary biology, designed to help biologists manage and analyze comparative data about organisms. Its emphasis is on phylogenetic analysis, but some of its modules concern population genetics, while others do non-phylogenetic multivariate analysis. Because it is modular, the analyses and management features available depend on the modules installed. Here is a brief overview of some of Mesquite’s features. See also a more complete outline of features, and the Mesquite Project Youtube channel, with instructional videos helping you learn Mesquite.
Despite Mesquite’s broad analytical capabilities, the developers of Mesquite find that we use Mesquite most often to provide a workflow of data editing, management, and processing. We will therefore begin there.
Definitely worth a look.
Share this:

Journal club light: skeptical of "Phylo SI: a new genome-wide approach for prokaryotic phylogeny"
Just reading this paper and thought I would start a new “section” here on my blog. Journal club light. Just some notes and quick comments.
Today I am selecting this paper: Phylo SI: a new genome-wide approach for prokaryotic phylogeny. It caught my eye because, well, I am interested in genome-wide phylogeny.
So I glanced at the paper’s abstract:
The evolutionary history of all life forms is usually represented as a vertical tree-like process. In prokaryotes, however, the vertical signal is partly obscured by the massive influence of horizontal gene transfer (HGT). The HGT creates widespread discordance between evolutionary histories of different genes as genomes become mosaics of gene histories. Thus, the Tree of Life (TOL) has been questioned as an appropriate representation of the evolution of prokaryotes. Nevertheless a common hypothesis is that prokaryotic evolution is primarily tree-like, and a routine effort is made to place new isolates in their appropriate location in the TOL. Moreover, it appears desirable to exploit non–tree-like evolutionary processes for the task of microbial classification. In this work, we present a novel technique that builds on the straightforward observation that gene order conservation (‘synteny’) decreases in time as a result of gene mobility. This is particularly true in prokaryotes, mainly due to HGT. Using a ‘synteny index’ (SI) that measures the average synteny between a pair of genomes, we developed the phylogenetic reconstruction tool ‘Phylo SI’. Phylo SI offers several attractive properties such as easy bootstrapping, high sensitivity in cases where phylogenetic signal is weak and computational efficiency. Phylo SI was tested both on simulated data and on two bacterial data sets and compared with two well-established phylogenetic methods. Phylo SI is particularly efficient on short evolutionary distances where synteny footprints remain detectable, whereas the nucleotide substitution signal is too weak for reliable sequence-based phylogenetic reconstruction. The method is publicly available at http://research.haifa.ac.il/ssagi/software/PhyloSI.zip.
And something continued to catch my eye there. It was the use of “gene order conservation” as the data for the phylogenetic analysis. Hmm. I am generally skeptical of most uses of gene order for inferring phylogeny that I have seen. Why? Well, because it seems to me that gene order is less likely to be a useful character than sequences in alignments (which is the standard for inferring phylogeny). Why do I feel this way? Well, for two main reasons:
1) Sequence alignments are robust. They have been used and used and used and shown to be quite powerful and useful (even though they are not perfect). The rich literature on alignments has shown where and when and how they are useful. And where and when and how they are not. And we have powerful, tested methods to use such alignments.
2) Gene order seems less likely to be robust. I am not saying it is not useful. But the literature I have seen suggests to me that gene order is more prone to convergent evolution than sequence. And gene order is more prone to enormous variation in rates and patterns of evolution. And gene order does not actually have a lot of characters to use compared to whole genome alignment based phylogenetics.
I could go on and on. There are many other reasons I prefer sequence alignments over gene order. But I am willing to consider that gene order could be more useful than I imagine. So I read on. And the first thing I did (which is almost always the first thing I do for new phylogenetic methods papers) was I looked at their phylogenetic results. And so off to Figure 9. And the results did little to convince me that their method was better than existing alignment based methods.

I am sure people cannot see this that well. But basically, I looked through the tree and there were just too many things that are inconsistent with trees that are very supported by lots of other data.
For example
which has in one clade species that almost certainly should not group together. In particular the presence of Neisseria in this group is very strange given that all other analysis put it in the Protebacteria and the Proteobacteria are found in other parts of the tree.
And there is another clade like this
With Francisella (also considered a Proteobacteria) in a clade with things from many other Phyla.
And then there is this one.
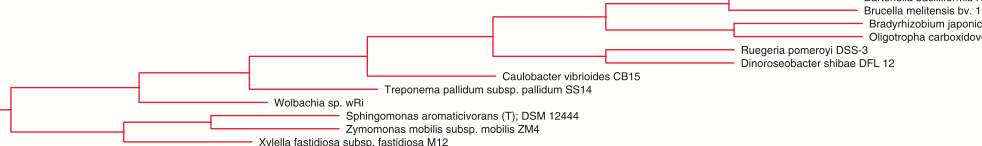
Which has gamma Proteobactera, alpha Proteobacteria, Spirochetes, and others all together in one clade.
I could go on. But this is journal club light. I just do not have time right now to dig much deeper. But on first look, I am certainly not overwhelmed with a desire to use gene order instead of sequence alignments to infer phylogenetic trees for bacteria. Again, I am not saying the method does not have its uses. It easily could be useful in many ways. But for inferring trees of all bacteria at once – does not seem to be the right thing.
Share this:

Rob Desalle Talk at UC Davis – Storification of Twitter Notes
New paper from some in the Eisen lab: phylogeny driven sequencing of cyanobacteria
(Cross post from my lab blog)

Quick post here. This paper came out a few months ago but it was not freely available so I did not write about it (it is in PNAS but was not published with the PNAS Open Option — not my choice – lead author did not choose that option and I was not really in the loop when that choice was made).
Improving the coverage of the cyanobacterial phylum using diversity-driven genome sequencing. [Proc Natl Acad Sci U S A. 2013] – PubMed – NCBI.
Anyway – it is now in Pubmed Central and at least freely available so I felt OK posting about it now. It is in a way a follow up to the “A phylogeny driven genomic encyclopedia of bacteria and archaea” paper (AKA GEBA) from 2009 with this paper a zooming in on the cyanobacteria.
Share this:

New paper from some in the Eisen lab: phylogeny driven sequencing of cyanobacteria
Quick post here. This paper came out a few months ago but it was not freely available so I did not write about it (it is in PNAS but was not published with the PNAS Open Option — not my choice – lead author did not choose that option and I was not really in the loop when that choice was made).
Improving the coverage of the cyanobacterial phylum using diversity-driven genome sequencing. [Proc Natl Acad Sci U S A. 2013] – PubMed – NCBI.
Anyway – it is now in Pubmed Central and at least freely available so I felt OK posting about it now. It is in a way a follow up to the “A phylogeny driven genomic encyclopedia of bacteria and archaea” paper (AKA GEBA) from 2009 with this paper a zooming in on the cyanobacteria.
Share this:
Twisted Tree of Life Award #16: Nature & Authors doing taxonomic alchemy converting an archaeon to a bacterium

Well, this is one of the bigger screw ups in terms of evolution I have seen at a major journal in a while. See the following paper in Nature: The catalytic mechanism for aerobic formation of methane by bacteria : Nature. The paper discusses some functions of “the ocean-dwelling bacterium Nitrosopumilus maritimus.” Some of what is reported in the paper is perhaps interesting (alas I do not have access). But painfully, there is one big big big big mistake – you see Nitrosopumilus maritimus is not a bacterium. It is an archaeon (see for example this paper on its genome).

I got pointed to this by Uri Gophna (in an email and in a comment on my blog)(all see this on Twitter) Sure – some people debate the structure of the tree of life. But I am pretty certain the authors here (Siddhesh S. Kamat, Howard J. Williams, Lawrence J. Dangott, Mrinmoy Chakrabarti & Frank M. Raushel) are not trying to make a statement about monophyly of bacteria or just what archaea are. They just made what seems to be a colossal screw up. And Nature not only let them, but added to it with things like their “Editors Summary”:
Novel bacterial biosynthesis of methane
Aerobic marine organisms produce significant quantities of the potent greenhouse gas methane, much of it via the cleavage of the highly unreactive carbon–phosphorus bonds of alkylphosphonates. In this study the authors explore the mechanism of PhnJ, an unusual radical S-adenosyl-L-methionine (SAM) enzyme that appears to use a cysteine-based thiyl radical to help catalyse the conversion of the alkylphosphonate substrate to methane and ribose-1,2-cyclic phosphate-5-phosphate. This reaction, not previously encountered in biological chemistry, establishes a novel mechanism for cleaving carbon–phosphorus bonds to form methane and phosphate via a covalent thiophosphate intermediate.
And for this taxonomic alchemy (converting an archaeon to a bacterium) I am awarding them and Nature my coveted “Twisted Tree of Life Award #16″.
UPDATE 5/28 7AM
I love the ad that came up while I was writing this post and searching for some information. I think Nature could use the services from this ad:

Share this:

The need for a phylogeny driven genomic encyclopedia of eukaryotes
Monday I gave a talk for the SMBE Eukaryotic Omics satellite meeting that has been going on at UC Davis. When Holly Bik, a post doc in my lab asked me to talk at the meeting, I said, basically “Well, OK, but I don’t really do much work on eukaryotes.” And then I came up with an idea – I could make my talk about how it might be good to have a better phylogenetic sampling of eukaryotic genome sequences. I have been a bit obsessed for many many years about phylogenetic sampling of genomes and, well, though I have avoided eukaryotes mostly in most of my genome sequencing work, I figured, I should still get on my soap box about how phylogenetic sampling is a good thing. So, well, I did. And I think we (i.e., the scientific community) really needs a better sampling of eukaryotic genomes.
I have posted my talk to Slideshare and I recorded audio of my talk in synch with the slides and posted that to Youtube. These are below.
I hereby am calling for those people interested in participating in such a phylogeny driven genomic encyclopedia of eukaryotes to make yourselves known. We NEED to do this.
Related posts
- The Tree of Life: Story Behind the Nature Paper on ‘A phylogeny driven genomic encyclopedia of bacteria and archaea…
- The Tree of Life: More coverage of the GEBA “Phylogeny Driven Genomic Encyclopedia paper …
- A phylogeny-driven genomic encyclopaedia of Bacteria and … – Nature
Share this:
If you are into microbial diversity and use R this may be worth checking out: phyloseq
Just got pointed (by the lead author) to this new paper: PLOS ONE: phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data
Abstract:
Background
The analysis of microbial communities through DNA sequencing brings many challenges: the integration of different types of data with methods from ecology, genetics, phylogenetics, multivariate statistics, visualization and testing. With the increased breadth of experimental designs now being pursued, project-specific statistical analyses are often needed, and these analyses are often difficult (or impossible) for peer researchers to independently reproduce. The vast majority of the requisite tools for performing these analyses reproducibly are already implemented in R and its extensions (packages), but with limited support for high throughput microbiome census data.
Results
Here we describe a software project, phyloseq, dedicated to the object-oriented representation and analysis of microbiome census data in R. It supports importing data from a variety of common formats, as well as many analysis techniques. These include calibration, filtering, subsetting, agglomeration, multi-table comparisons, diversity analysis, parallelized Fast UniFrac, ordination methods, and production of publication-quality graphics; all in a manner that is easy to document, share, and modify. We show how to apply functions from other R packages to phyloseq-represented data, illustrating the availability of a large number of open source analysis techniques. We discuss the use of phyloseq with tools for reproducible research, a practice common in other fields but still rare in the analysis of highly parallel microbiome census data. We have made available all of the materials necessary to completely reproduce the analysis and figures included in this article, an example of best practices for reproducible research.
Conclusions
The phyloseq project for R is a new open-source software package, freely available on the web from both GitHub and Bioconductor.
Seems similar in some ways to the WATERs Kepler Workflow that we released a few years ago. Anyway – if you use R and are into microbial diversity studies this may be worth checking out. As a bonus – it has a strong emphasis on reproducibility – which is a good thing.
