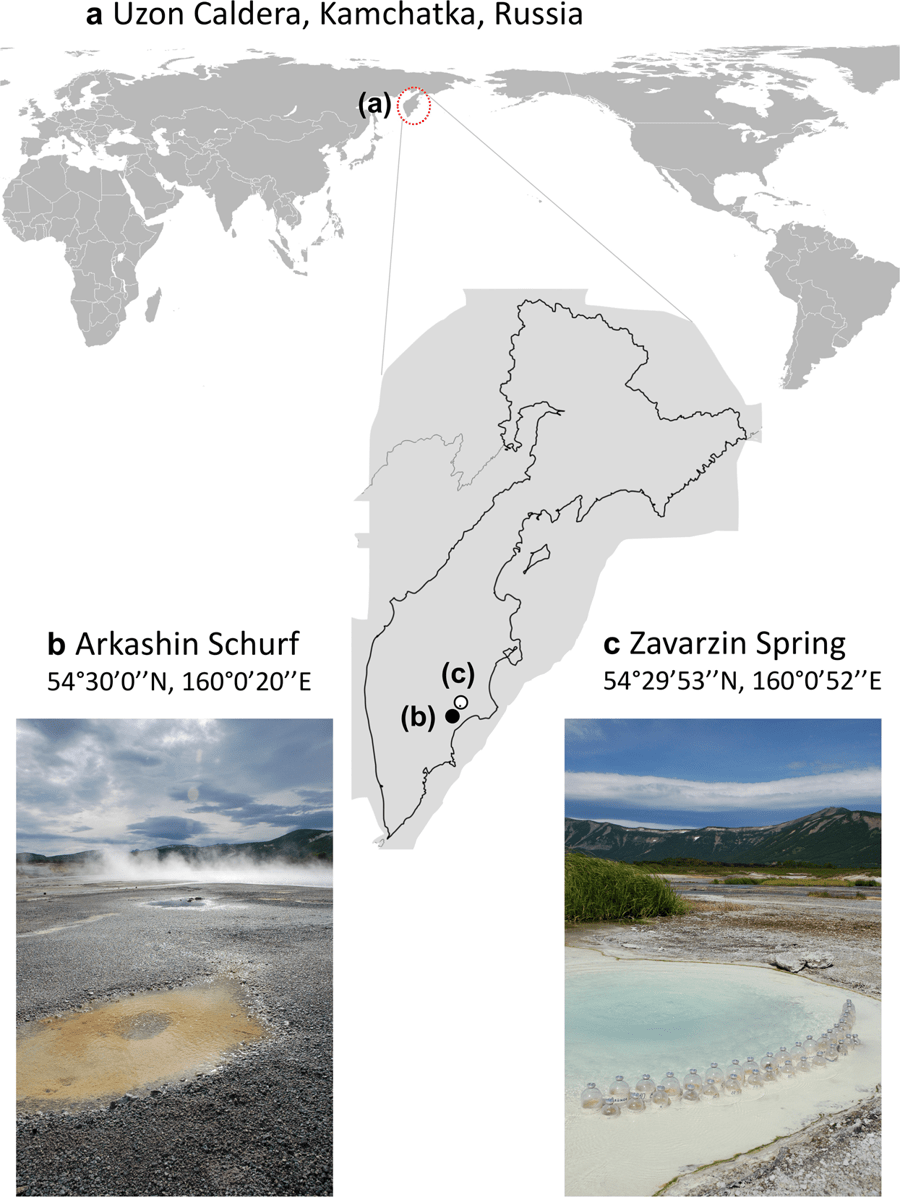
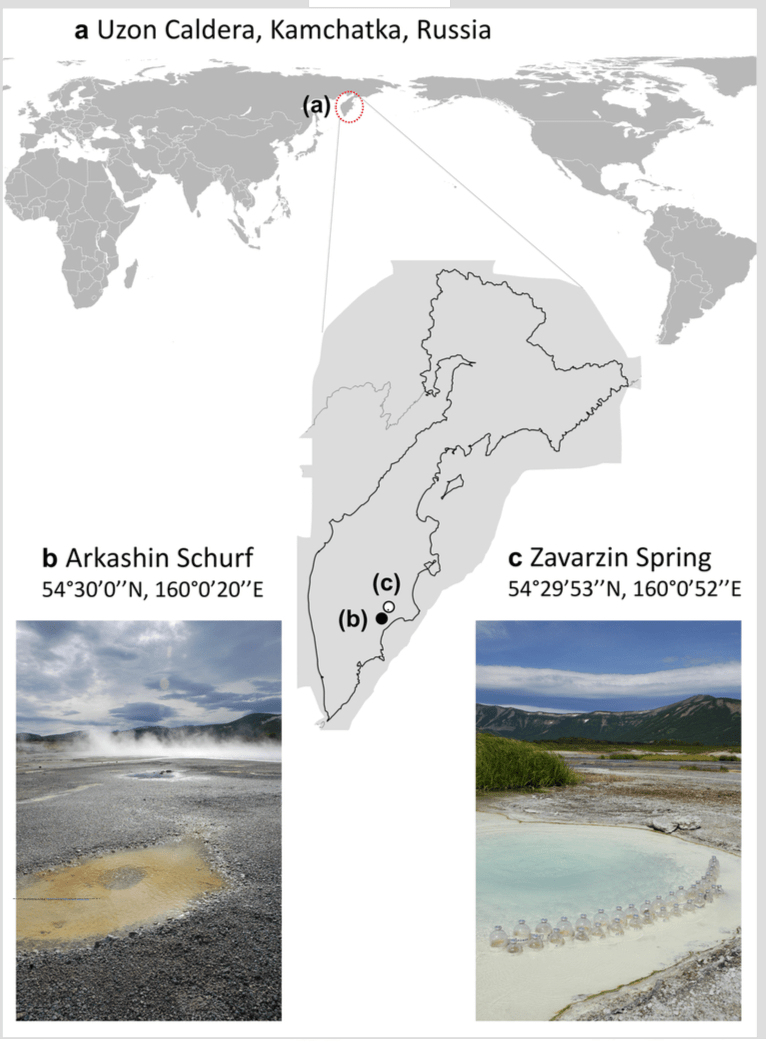
A new paper just out from the lab:
Laetitia G. E. Wilkins & Cassandra L. Ettinger (co-1st authors), Guillaume Jospin & Jonathan A. Eisen. Metagenome-assembled genomes provide new insight into the microbial diversity of two thermal pools in Kamchatka, Russia. Scientific Reports. volume 9, Article number: 3059
This was truly a remarkable piece of work led by Laetitia G. E. Wilkins & Cassandra L. Ettinger who are co-first authors on the paper. Also contributing to the work was Guillaume Jospin, the lab bioinformatics guru. I will write a much longer “story behind the paper” about this in the next week or two but wanted to give a brief summary here.
This paper had its beginnings just after September 11, 2001. Yes. that September 11. We had a meeting planned in September in Yellowstone National Park that had to be delayed due to 9/11. The meeting then happened in October. The meeting was between a group of US researchers and a group of Russian researchers to plan an NSF proposal on comparing the microbes living in hot springs in Yellowstone with those living in hot springs in Kamchatka. The meeting went well and eventually we got a grant from NSF’s Biocomplexity program for this work.
At the time I was at TIGR (The Institute for Genomic Research) and I had a minor role in the grant – I was supposed to do coordinate some metagenomic sequencing of samples from Kamchatka. We got some samples from Juergen Wiegel from U. Georgia and did a small amount of Sanger sequencing of them but this was right around the time I moved to UC Davis and right around the time that TIGR kind of dissolved. We did not end up writing a paper on that small amount of Sanger sequencing. Years later, Russell Neches in my lab got interested in this project and went to Kamchatka with Frank Robb and others on a collecting trip. See some details about this trip and plans back then in this blog post. Russell then coordinated some Illumina sequencing of the same DNA samples we had done Sanger sequencing for at TIGR. And Russell did some preliminary analysis of the samples but did not end up writing up a paper on it.
Fast forward again to a few years ago and we decided in my lab to try and rescue orphan data and try to at least get the data into the public domain and, if it was of interest to someone, write a paper on the data. We thus created some “Reboot” channels within the lab Slack site and Laetitia Wilkins and Cassie Ettinger decided to try and do something with the Kamchatka data. And they did. This is what led to this paper.
A few other notes I would like to make here. This paper certainly is a testament to the remarkable work of Cassie Ettinger and Laetitia Wilkins as well as Guillaume Jospin who helped them with some of the informatics. I am really proud to have them all in my lab. In addition, this paper is a culmination of contributions of all sorts of people. We tried to acknowledge many of them in the Acknowledgement section of the paper and I am posting that below for the record here.
We would like to thank Russell Neches (ORCID: 0000-0002-2055-8381) for the use of photographs taken on a trip to Kamchatka, Russia in 2012. We also would like to thank Elizabeth A. Burgess and Juergen Wiegel for providing JAE with the DNA used here. We are also grateful to Christopher Brown (ORCID: 0000-0002-7758-6447) and Laura Hug (ORCID: 0000-0001-5974-9428) for their help getting access to the genomes used in Figure 2. A special thank you goes to Alexandros Stamatakis (ORCID: 0000-0003-0353-0691), Wayne Pfeiffer and Mark Miller for offering their help with getting the CIPRES Science Gateway server to run RAxML-HPC2 v.8 on XSEDE. We also thank two anonymous reviewers for comments on earlier versions of this manuscript. A. Murat Eren (ORCID: 0000-0001-9013-4827) provided constructive feedback on the publicly available preprint version of this article. LGEW was supported by a fellowship of the Swiss National Science Foundation P2LAP3_164915. Funding for the sequencing at TIGR was provided by a a subcontract to JAE for a grant from the National Science Foundation (MCB-MO 0238407). Funding for some of the work on this project was also provided by grant from the Gordon and Betty Moore Foundation (GBMF5603) to JAE.