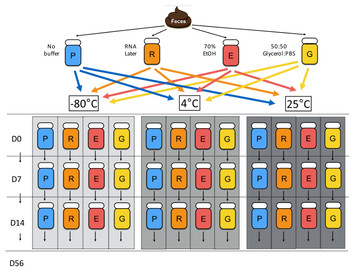
New paper out from the lab. This is from a collaboration with Stan Marks in the Vet School at UC Davis. The work was led by Katti Horng with assistance from Holly Ganz.
Citation: (2018) Effects of preservation method on canine (Canis lupus familiaris) fecal microbiota. PeerJ 6:e4827https://doi.org/10.7717/peerj.4827
Abstract: Studies involving gut microbiome analysis play an increasing role in the evaluation of health and disease in humans and animals alike. Fecal sampling methods for DNA preservation in laboratory, clinical, and field settings can greatly influence inferences of microbial composition and diversity, but are often inconsistent and under-investigated between studies. Many laboratories have utilized either temperature control or preservation buffers for optimization of DNA preservation, but few studies have evaluated the effects of combining both methods to preserve fecal microbiota. To determine the optimal method for fecal DNA preservation, we collected fecal samples from one canine donor and stored aliquots in RNAlater, 70% ethanol, 50:50 glycerol:PBS, or without buffer at 25 °C, 4 °C, and −80 °C. Fecal DNA was extracted, quantified, and 16S rRNA gene analysis performed on Days 0, 7, 14, and 56 to evaluate changes in DNA concentration, purity, and bacterial diversity and composition over time. We detected overall effects on bacterial community of storage buffer (F-value = 6.87, DF = 3, P < 0.001), storage temperature (F-value=1.77, DF = 3, P = 0.037), and duration of sample storage (F-value = 3.68, DF = 3, P < 0.001). Changes in bacterial composition were observed in samples stored in −80 °C without buffer, a commonly used method for fecal DNA storage, suggesting that simply freezing samples may be suboptimal for bacterial analysis. Fecal preservation with 70% ethanol and RNAlater closely resembled that of fresh samples, though RNAlater yielded significantly lower DNA concentrations (DF = 8.57, P < 0.001). Although bacterial composition varied with temperature and buffer storage, 70% ethanol was the best method for preserving bacterial DNA in canine feces, yielding the highest DNA concentration and minimal changes in bacterial diversity and composition. The differences observed between samples highlight the need to consider optimized post-collection methods in microbiome research.
Source: Effects of preservation method on canine (Canis lupus familiaris) fecal microbiota