
//storify.com/phylogenomics/softciteucd/embed?border=false//storify.com/phylogenomics/softciteucd.js?border=false[View the story “Software for Science: Getting Credit for Code #SoftCiteUCD” on Storify]
Tag: software

Mesquite "A modular system for evolutionary analysis" v3.0 released from Team Maddison
Just found out about this on Facebook via Rod Page: Mesquite V3.0 has been released. Mesquite is from Team Maddison (Wayne and David). I have been using their software since 1987 when I took Stephen Jay Gould’s course at Harvard and they were TAs for the course demoing an early version of MacClade. Lots of nice features and it is available in Mac, Unix/Linux, and Windows versions. They describe “What Mesquite Does” on their Wikispaces site in the following way:
Mesquite is software for evolutionary biology, designed to help biologists manage and analyze comparative data about organisms. Its emphasis is on phylogenetic analysis, but some of its modules concern population genetics, while others do non-phylogenetic multivariate analysis. Because it is modular, the analyses and management features available depend on the modules installed. Here is a brief overview of some of Mesquite’s features. See also a more complete outline of features, and the Mesquite Project Youtube channel, with instructional videos helping you learn Mesquite.
Despite Mesquite’s broad analytical capabilities, the developers of Mesquite find that we use Mesquite most often to provide a workflow of data editing, management, and processing. We will therefore begin there.
Definitely worth a look.
Share this:

New EisenLab paper: PhyloSift: phylogenetic analysis of genomes and metagenomes [PeerJ]
New paper from people in the Eisen lab (and some others): PhyloSift: phylogenetic analysis of genomes and metagenomes [PeerJ]. This project was coordinated by Aaron Darling, who was a Project Scientist in my lab and is now a Professor at the University of Technology Sydney. Also involved were Holly Bik (post doc in the lab), Guillaume Jospin (Bioinformatics Engineer in the lab), Eric Lowe (was a UC Davis undergrad working in the lab) and Erick Matsen (from the FHCRC). This work was supported by a grant from the Department of Homeland Security.
Abstract:
Like all organisms on the planet, environmental microbes are subject to the forces of molecular evolution. Metagenomic sequencing provides a means to access the DNA sequence of uncultured microbes. By combining DNA sequencing of microbial communities with evolutionary modeling and phylogenetic analysis we might obtain new insights into microbiology and also provide a basis for practical tools such as forensic pathogen detection.
In this work we present an approach to leverage phylogenetic analysis of metagenomic sequence data to conduct several types of analysis. First, we present a method to conduct phylogeny-driven Bayesian hypothesis tests for the presence of an organism in a sample. Second, we present a means to compare community structure across a collection of many samples and develop direct associations between the abundance of certain organisms and sample metadata. Third, we apply new tools to analyze the phylogenetic diversity of microbial communities and again demonstrate how this can be associated to sample metadata.
These analyses are implemented in an open source software pipeline called PhyloSift. As a pipeline, PhyloSift incorporates several other programs including LAST, HMMER, and pplacer to automate phylogenetic analysis of protein coding and RNA sequences in metagenomic datasets generated by modern sequencing platforms (e.g., Illumina, 454).
For more about Phylosift see
- gjospin/PhyloSift · GitHub
- PhyloSift | mining the global metagenome
- PhyloSift | Jonathan Eisen’s Lab
- PhyloSift (PhyloSift) on Twitter
- PhyloSift – Google Groups
Share this:
Thanks to Software Carpentry (@swcarpentry) for coming to #UCDavis
Quick post here. Jenna Lang in my lab has a post at microBEnet about the recent workshop that the Software Carpentry folks ran at UC Davis: Software Carpentry comes to UC Davis! | microBEnet: The microbiology of the Built Environment network. It was a major success. For those who don’t know Software Carpentry’s mission is is to build basic computing skills among researchers. From their web site:
Software Carpentry helps researchers be more productive by teaching them basic computing skills. We run boot camps at dozens of sites around the world, and also provide open access material online for self-paced instruction. The benefits are more reliable results and higher productivity: a day a week is common, and a ten-fold improvement isn’t rare.
A great idea and done really well. Others out there should consider hosting or attending one of their Boot Camps and checking out their materials on their web site. See for example their videos and their reading list and their lessons. They really do great things …
Share this:
Guest post on "CHANCE" ChIP-seq QC and validation software
Guest post by Aaron Diaz from UCSF on a software package called CHANCE which is for ChIP-seq analyses. Aaron wrote to me telling me about the software and asking if I would consider writing about it on my blog. Not really the normal topic of my blog but it is open source and published in an open access journal and is genomicy and bioinformaticy in nature. So I wrote back inviting him to write about it. Here is his post:
CHANCE: A comprehensive and easy-to-use graphical software for ChIP-seq quality control and validation

Our recent paper presents CHANCE a user-friendly software for ChIP-seq QC and protocol optimization. Our user-friendly graphical software quickly estimates the strength and quality of immunoprecipitations, identifies biases, compares the user’s data with ENCODE’s large collection of published datasets, performs multi-sample normalization, checks against qPCR-validated control regions, and produces publication ready graphical reports. CHANCE can be downloaded here.
 |
|
An overview of ChIP-seq: cross-
linked chromatin is sheared,
enriched for a transcription factor
or epigenetic mark of interest
using an antibody, purified and
sequenced.
|
Chromatin immunoprecipitation followed by high throughput sequencing (ChIP-seq) is a powerful tool for constructing genome wide maps of epigenetic modifications and transcription factor binding sites. Although this technology enables the study of transcriptional regulation with unprecedented scale and throughput interpreting the resulting data and knowing when to trust the data can be difficult. Also, when things go wrong it is hard to know where to start when troubleshooting. CHANCE provides a variety of tests to help debug library preparation protocols.
One of the primary uses of CHANCE is to check the strength of the IP. CHANCE produces a summary statement which will give you an estimate of the percentage of the IP reads which map DNA fragments pulled down by the antibody used for the ChIP. In addition to the size of this signal component within the IP CHANCE reports the fraction of the genome these signal reads cover, as well as the statistical significance of the genome wide percentage enrichment relative to control in the form of a q-value (positive false discovery rate). CHANCE has been trained on CHIP-seq experiments from the ENCODE repository by making over 10,000 Input to IP and Input to replicate Input comparisons. The q-value reported gives then the fraction of comparisons between Input sample techinical replicates that report an enrichment for signal in one sample compared to another equal to the user provided sample or greater. CHANCE identifies insufficient sequencing depth, PCR amplification bias in library preparation, and batch effects.
CHANCE identifies biases in sequence content and quality, as well as cell-type and laboratory-dependent biases in read density. Read-density bias reduces the statistical power to distinguish subtle but real enrichment from background noise. CHANCE visualizes base-call quality and nucleotide frequency with heat maps. Furthermore, efficient techniques borrowed from signal processing uncover biases in read density caused by sonication, chemical digestion, and library preparation.
 |
| A typical IP enrichment report. |
CHANCE cross-validates enrichment with previous ChIP-qPCR results. Experimentalists frequently use ChIP-qPCR to check the enrichment of positive control regions and the background level of negative control regions in their IP DNA relative to Input DNA. It is thus important to verify whether those select regions originally checked with PCR are captured correctly in the sequencing data. CHANCE’s spot-validation tool provides a fast way to perform this verification. CHANCE also compares enrichment in the user’s experiment with enrichment in a large collection of experiments from public ChIP-seq databases.
 |
| CHANCE has a user friendly graphical interface. |
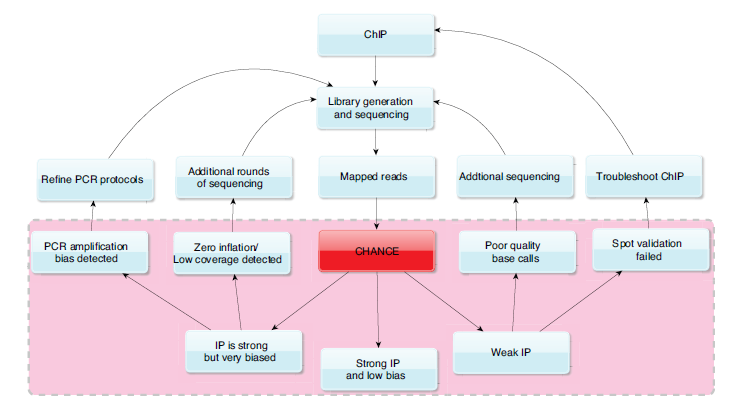 |
| How CHANCE might be used to provide feedback on protocol optimization. |
Share this:

New publication from members of my lab (e.g., @ryneches) & lab of Marc Facciotti on ChIP-seq based mapping of archaeal transcription factors
New publication from members of my lab and the lab of Marc Facciotti on a workflow for ChIP-seq based mapping of archaeal transcription factors. The paper includes a description of new software from Russell Neches in my lab called pique for peak calling.
See: A workflow for genome-wide mapping of archaeal transcription factors with ChIP-seq
Russell’s pique software is available on github here: https://github.com/ryneches/pique.
 |
|
The Pique software package processes ChIP-seq coverage data to predict protein-binding sites. Strand-specific coverage data are output as tracks for the Gaggle Genome Browser, and putative-binding sites (peaks) are output as ‘bookmark files’. (A) Screenshot of data browsing in the Gaggle Genome Browser. Green box outlines the navigation window for clicking through bookmarks of predicted binding sites. Details of each site can be displayed (inset). The Gaggle toolbar (shown with black arrow) can be used to broadcast selected data to other ‘geese’ in the gaggle package, programs such as R, cytoscape, BLAST or KEGG. (B) Schematic overview of bioinformatics workflow.
|
![]() Wilbanks, E., Larsen, D., Neches, R., Yao, A., Wu, C., Kjolby, R., & Facciotti, M. (2012). A workflow for genome-wide mapping of archaeal transcription factors with ChIP-seq Nucleic Acids Research DOI: 10.1093/nar/gks063
Wilbanks, E., Larsen, D., Neches, R., Yao, A., Wu, C., Kjolby, R., & Facciotti, M. (2012). A workflow for genome-wide mapping of archaeal transcription factors with ChIP-seq Nucleic Acids Research DOI: 10.1093/nar/gks063
Share this:

New openaccess paper from my lab on "Zorro" software for automated masking of sequence alignments
A new Open Access paper from my lab was just published in PLoS One: Accounting For Alignment Uncertainty in Phylogenomics. Wu M, Chatterji S, Eisen JA (2012) Accounting For Alignment Uncertainty in Phylogenomics. PLoS ONE 7(1): e30288. doi:10.1371/journal.pone.0030288
The paper describes the software “Zorro” which is used for automated “masking” of sequence alignments. Basically, if you have a multiple sequence alignment you would like to use to infer a phylogenetic tree, in some cases it is desirable to block out regions of the alignment that are not reliable. This blocking is called “masking.”
Masking is thought by many to be important because sequence alignments are in essence a hypothesis about the common ancestry of specific residues in different genes/proteins/regions of the genome. This “positional homology” is not always easy to assign and for regions where positional homology is ambiguous it may be better to ignore such regions when inferring phylogenetic trees from alignments.
Historically, masking has been done by hand/eye looking for columns in a multiple sequence alignment that seem to have issues and then either eliminating those columns or giving them a lower weight and using a weighting scheme in the phylogenetic analysis.
What Zorro does is it removes much of the subjectivity of this process and generates automated masking patterns for sequence alignments. It does this by assigning confidence scores to each column in a multiple seqeunce alignment. These scores can then be used to account for alignment accuracy in phylogenetic inference pipelines.
The software is available at Sourceforge: ZORRO – probabilistic masking for phylogenetics. It was written primarily by Martin Wu (who is now a Professor at the University of Virginia) and Sourav Chatterji with a little help here and there from Aaron Darling I think. The development of Zorro was part of my “iSEEM” project that was supported by the Gordon and Betty Moore Foundation.
In the interest of sharing, since the paper is fully open access, I am posting it here below the fold. UPDATE 2/9 – decided to remove this since it got in the way of getting to the comments …
Share this:
One old, one new – a few phylogeny papers worth checking out
Just a quick one here. A few days ago in my lab we were discussing some challenges with doing phylogenetic diversity (PD) measurements in very very large phylogenetic trees. PD is a measure of total branch length in a phylogenetic tree for a group of taxa … and we use it for many purposes.
For many of our applications we have been using an algorithm described by Mike Steele “Phylogenetic diversity and the Greedy Algorithm“. But alas, is is not keeping up with the massive tree sets we are dealing with. Fortunately Aaron Darling in my lab found a alternative paper with a perfect sounding title for us: Phylogenetic Diversity within Seconds from Minh, Klaere, and von Haeseler. This seems like it will do the trick. I note – Kudos to Systematic Biology for making some older papers freely available. Not sure of their general policies on this but good to see.
Anyway – back to the grind …
Share this:
Draft post cleanup #11: Tree Hugging
Yet another post in my “draft blog post cleanup” series. Here is #11 from September.
Just a quick one. In August a nice review paper came out on phylogenetic analysis software: Learning to Become a Tree Hugger | The Scientist. By Amy Maxmen it is a “A guide to free software for constructing and assessing species relationships”. Definitely worth checking out.
Among the key links & tools discussed:
- Clustal
- RAxML
- MrBayes
- BEAST
- TNT
- BEAST user group
- FigTree
- Mesquite
- Dendroscope
- Paloverde
- GeoPhylo
- MUSCLE
- MAFFT
- T-Coffee
- SeaView
- Phylogeny.fr
- European Bioinformatics Institute
- CIPRES
- Evolutionary Analysis Mesquite
- Biodiverse
- Bayes Traits
- Lagrange
Share this:
New paper from my lab (& the Facciotti lab): Mauve Assembly Metrics #Halophiles #Genomics
Just a quick post here. A new paper from my lab has come out in Bioinformatics. The paper is relatively simple. Titled “Mauve Assembly Metrics” it reports work of Aaron Darling and Andrew Tritt (with some minor contributions from me and Marc Facciotti). Aaron wrote the program Mauve when he was a student in Nicole Perna’s lab at Wisconsin: Mauve: multiple alignment of conserved genomic sequence with rearrangements. Over the years he (and others) have continued to develop the program and written a few papers too including for example, the development of progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. This new paper reports basically a system/scripts to measure assembly quality. Here is the abstract:
High throughput DNA sequencing technologies have spurred the development of numerous novel methods for genome assembly. With few exceptions, these algorithms are heuristic and require one or more parameters to be manually set by the user. One approach to parameter tuning involves assembling data from an organism with an available high quality reference genome, and measuring assembly accuracy using some metrics. We developed a system to measure assembly quality under several scoring metrics, and to compare assembly quality across a variety of assemblers, sequence data types, and parameter choices. When used in conjunction with training data such as a high quality reference genome and sequence reads from the same organism, our program can be used to manually identify an optimal sequencing and assembly strategy for de novo sequencing of related organisms.
Check out the paper: Mauve Assembly Metrics. Download the scripts/code http://ngopt.googlecode.com and Mauve and play around and let me know what you think.
Note this paper was supported by a grant from the National Science Foundation (ER 0949453). That grant is focused on comparative genomics (sequencing and analysis) of halophlic archaea. Stay tuned for more on that project as we are writing up a series of papers ….
Some related links:
