Guest post by Aaron Diaz from UCSF on a software package called CHANCE which is for ChIP-seq analyses. Aaron wrote to me telling me about the software and asking if I would consider writing about it on my blog. Not really the normal topic of my blog but it is open source and published in an open access journal and is genomicy and bioinformaticy in nature. So I wrote back inviting him to write about it. Here is his post:
CHANCE: A comprehensive and easy-to-use graphical software for ChIP-seq quality control and validation

Our
recent paper presents CHANCE a user-friendly software for ChIP-seq QC and protocol optimization. Our user-friendly graphical software quickly estimates the strength and quality of immunoprecipitations, identifies biases, compares the user’s data with ENCODE’s large collection of published datasets, performs multi-sample normalization, checks against qPCR-validated control regions, and produces publication ready graphical reports. CHANCE can be downloaded
here.
 |
|
An overview of ChIP-seq: cross-
linked chromatin is sheared,
enriched for a transcription factor
or epigenetic mark of interest
using an antibody, purified and
sequenced.
|
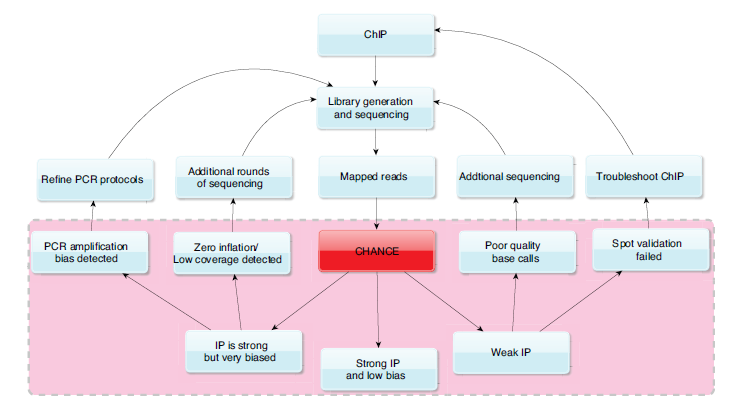
Chromatin immunoprecipitation followed by high throughput sequencing (ChIP-seq) is a powerful tool for constructing genome wide maps of epigenetic modifications and transcription factor binding sites. Although this technology enables the study of transcriptional regulation with unprecedented scale and throughput interpreting the resulting data and knowing when to trust the data can be difficult. Also, when things go wrong it is hard to know where to start when troubleshooting. CHANCE provides a variety of tests to help debug library preparation protocols.
One of the primary uses of CHANCE is to check the strength of the IP. CHANCE produces a summary statement which will give you an estimate of the percentage of the IP reads which map DNA fragments pulled down by the antibody used for the ChIP. In addition to the size of this signal component within the IP CHANCE reports the fraction of the genome these signal reads cover, as well as the statistical significance of the genome wide percentage enrichment relative to control in the form of a q-value (positive false discovery rate). CHANCE has been trained on CHIP-seq experiments from the ENCODE repository by making over 10,000 Input to IP and Input to replicate Input comparisons. The q-value reported gives then the fraction of comparisons between Input sample techinical replicates that report an enrichment for signal in one sample compared to another equal to the user provided sample or greater. CHANCE identifies insufficient sequencing depth, PCR amplification bias in library preparation, and batch effects.
CHANCE identifies biases in sequence content and quality, as well as cell-type and laboratory-dependent biases in read density. Read-density bias reduces the statistical power to distinguish subtle but real enrichment from background noise. CHANCE visualizes base-call quality and nucleotide frequency with heat maps. Furthermore, efficient techniques borrowed from signal processing uncover biases in read density caused by sonication, chemical digestion, and library preparation.
 |
| A typical IP enrichment report. |
CHANCE cross-validates enrichment with previous ChIP-qPCR results. Experimentalists frequently use ChIP-qPCR to check the enrichment of positive control regions and the background level of negative control regions in their IP DNA relative to Input DNA. It is thus important to verify whether those select regions originally checked with PCR are captured correctly in the sequencing data. CHANCE’s spot-validation tool provides a fast way to perform this verification. CHANCE also compares enrichment in the user’s experiment with enrichment in a large collection of experiments from public ChIP-seq databases.
 |
| CHANCE has a user friendly graphical interface. |
 |
| How CHANCE might be used to provide feedback on protocol optimization. |