FridayGBSF 1005
Noon
Dr. Michael Fischbah of UCSF
"Insights from a global view of secondary metabolism: Small molecules from the human microbiota”

FridayGBSF 1005
Noon
Dr. Michael Fischbah of UCSF
"Insights from a global view of secondary metabolism: Small molecules from the human microbiota”
“Exploring the impact of noncoding genetic variation in humans”
Hundreds of genome-wide association studies (GWAS) have found that the vast majority of human genetic variation impacting complex traits occurs outside of protein-coding regions, yet our understanding of these noncoding variants is still quite poor. I will describe our recent efforts to explore the impact of these functional variants. We have developed new experimental approaches to detect which variants affect key components of transcriptional regulation such as transcription factor binding, DNA methylation, and histone modification. We are also developing new computational methods for detecting natural selection in noncoding regions, revealing a dominant role of cis-regulatory changes in recent human adaptation.
Speaker: Hunter Fraser
Assistant Professor
Stanford University
Monday, May 18, 2015
4:10-5:00 PM
1022 Life Sciences
Please join us for Alex Csiszar (Harvard University) speaking on "The Scientific Journal: A Political History"
Wednesday, May 13, 2015, 12:00 pm – 1:30 pm
2nd Floor Instruction Room, Shields Library
Lunch will be served – Please RSVP at this link if you plan to attend http://goo.gl/forms/4D9nm6NXps
Scientific journals are expected to do a lot of different things. They are often identified with both the cumulative and the present state of knowledge possessed by scientific communities. Journals are supposed to be public enough that any interested reader might access them; yet at the same time they are rigorously exclusive. To publish in a particular set of journals is to be deemed an expert in the corresponding scientific field. When questions arise as to what scientific consensus is on some matter of concern, governmental bodies, the public, and journalists routinely look to the reputable journal literature dealing with that subject. The list of a researcher’s papers is a unit by which careers are measured and a dominant factor in decisions about hiring, tenure, and grants. Scientific journals are both permanent archive and breaking news, both complete record and painstaking selection, both public forum and the esoteric domain of experts.
This talk will explore how and why this improbable state of affairs came into being over the course of the nineteenth century. The shift whereby the authority of science came to be vested increasingly in serialized print did not come about through any deliberate decision taken by scientists based on the fitness of the periodical press to play this role. Far from emerging out of a timeless need for a secure communications medium and format, the ascendancy of the scientific journal occurred as European scientific elites sought to establish their collective legitimacy to speak authoritatively about nature following the political upheavals of the Napoleonic Wars. Since that time, the scientific journal has been a nodal point where expert cultures of credibility have intersected, uneasily, with public criteria of accountability.
Alex Csiszar is an Assistant Professor of the History of Science at Harvard University. His research concerns the history of scientific authorship, publishing, refereeing practices, and information management from the French Revolution to the twentieth century.
Well, this is one of the worst microbiome news stories in a long time: Fast food kills gut bacteria that can keep you slim, book claims. So many things wrong with it I don’t even know where to go. Here are nine:
1. The original headline: “Fast food kills gut bacteria that can keep you slim, study finds”

Here is the Tweet
Fast food kills gut bacteria that can keep you slim, study finds http://t.co/3fPIhJTuAB | @newshour
— PBS (@PBS) May 11, 2015
//platform.twitter.com/widgets.js
2. The correction:
is just completely lame and they should, as the New York Times does when it makes a correction, say what it used to say before they changed it
3. The sentence with the reference to Rob Knight is just bad reporting #1
Here is the quote:
Previous studies made similar findings: Professor Rob Knight of the University of Colorado Boulder, who collaborates with Spector, famously showed that transferring gut bacteria from obese humans to mice could make the rodents gain weight.
First of all – the paper they link to does include Rob Knight as a co-author, but the corresponding and senior author is Jeffrey Gordon and Rob is fourth to last (mind you I love Rob and his work, but in this case, saying this is something Rob showed without mentioning Gordon is just not right).
4 . The sentence with the reference to Rob Knight is just bad reporting #2
What the *$*$# does “famously showed” mean? Really. What does it mean?
5 . The sentence with the reference to Rob Knight is just bad reporting #3
The statement “Previous studies made similar findings” is just so incredibly misleading. It seems to be referring back to the previous sentences:
“What is emerging is that changes in our gut microbe community , or microbiome, are likely to be responsible for much of our obesity epidemic, and consequences like diabetes, cancer and heart disease,” he said. “It is clear that the more diverse your diet, the more diverse your microbes and the better your health at any age.”
This is just completely overblown. The more diverse your diet the better your health at any age? Oh #FFS that is just not based on any science. And the “likely responsible for” is silly too.
6 . The sentence with the reference to Rob Knight is just bad reporting #4
Why exactly tell us he is collaborating with Rob Knight? So some of Rob’s good work rubs off? I mean, Spector may do some fine work (and he has done some really good stuff). But casually mentioning he collaborates with Knight who famously showed something (when actually it was more Jeff Gordon’s work) which did not actually show what the article implies it showed. Aaaaaaaaaaarg.
7. Good news.
Spector’s book claims that the diversity of microbes in the human body has decreased almost a third over the last century. But there’s also good news: Foods like dark chocolate, garlic, coffee and Belgian beer may help increase gut microbes.
Really? Thinking about microbes MAY also increase gut microbes. And so might listening to NPR. Not something worth reporting here.
8. This sentence
This discovery suggested to his father that many cases of obesity may not simply be due to overeating.
That is right. Looking at his son’s poop and the microbes in it is the key to knowing that obesity might actually be fuc$*@#@ complex and not only caused by overeating. Oh, that and 100 years of epidemiology and research.
9. This sentence
“Once on the diet I rapidly lost 1,300 species of bacteria and my gut was dominated by a different group called bacteroidetes. The implication is that the McDonald’s diet killed 1,300 of my gut species,” he said.
Sorry but that is NOT the implication.
UPDATE 1: May 11, 2015. 8:00 PM
Thanks to a Tweet from Jennifer Gunter I changed the title of my post from ” 9 things horribly wrong with Newshour story on fast food and microbiomes” to “9 things PBS Newshour famously gets horrible wrong in story on fast food and microbiomes”
@PBS famously showed bad science reporting https://t.co/8VuGq5njWU
— Jennifer Gunter (@DrJenGunter) May 12, 2015
//platform.twitter.com/widgets.js
With apologies I don’t have time right now to tease apart all the details on these meetings. But, yuck. Cell Symposia have a big and persistent problem with gender balance of speakers. See the Storify below:



Just got this via the email tubes:
FuturePhy is an NSF-sponsored, three-year program of conferences, workshops and hackathons on the Tree of Life. The project aims to promote novel, integrative data analyses and visualization, interdisciplinary syntheses of phylogenetic sciences, and cross-cutting uses of phylogenetics to develop and address new research questions and applications.
The first phase of this mission is critical: to bring together a broad community of people from diverse backgrounds who are active in phylogenetics research, who use the tree of life in research or education, who will benefit in applied or practical ways from a comprehensive tree of life, or who come from a background that offers new perspectives on defining, addressing or transcending key challenges in phylogenetics.
Help accelerate progress in all aspects of phylogenetics research by joining FuturePhy today. Diverse opportunities will be available to attend FuturePhy sessions in person or virtually, and to link FuturePhy to existing projects and initiatives.
We invite you to participate in the project in several ways:
1. Register on http://futurephy.org Scientists from all aspects of the phylogenetic sciences, educators, members of the tree-using community, and others interested in phylogenetics are welcome.
2. Contribute to the discussion forum on http://futurephy.org This is the best way to log your interest and contribute ideas.
3. See our main themes, comment on them and take the survey to rank them: https://www.surveymonkey.com/s/8CWXMRF
4. Email contact with ideas or comments
5. Tweet to the FuturePhy community: @FuturePhy
6. Comment in the FuturePhy thread on http://phylobabble.org
Special Seminar: Entomology, Bee biology
"Pesticides, parasites and pollinators: the impacts of environmental stressors on bees"
Prof Nigel Raine Univ of Guelph
Date: Friday May 8
Time: 1 PM
RM: 366 Briggs Hall
Recently I received and email from Rachid Ounit pointing me to a new open access paper he had on a metagenomics analysis tool called CLARK. I asked him if he would be willing to write a guest post about it and, well, he did. Here is it:
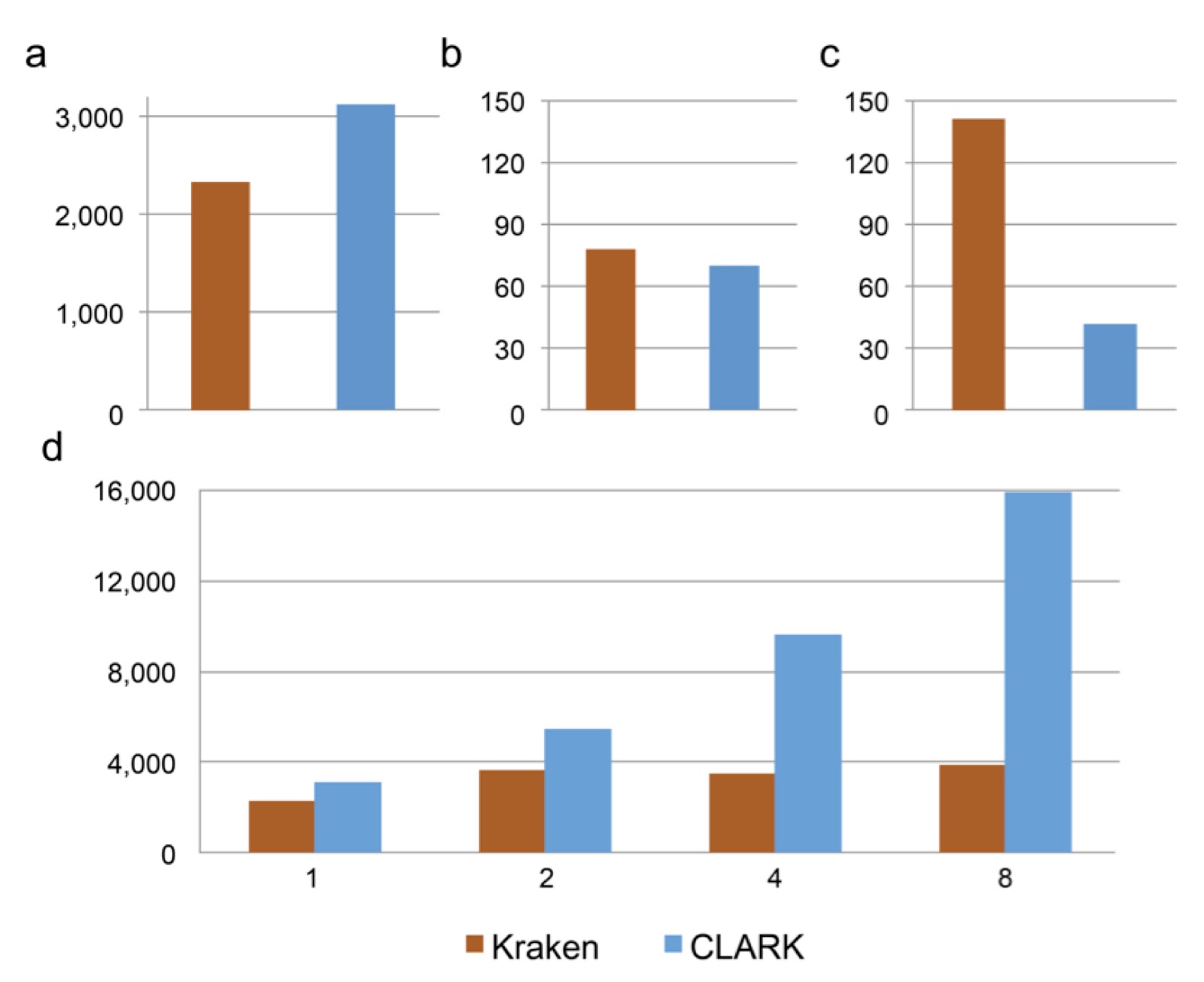 |
| Performance of Kraken (v0.10.4-beta) and CLARK (v1.0) for the classification of a metagenome sample of 10,000 reads (average reads length 92bp). a) The classification speed (in 103 reads per minute) in default mode. b) RAM usage (in GB) for the classification. c) Disk space (in GB) required for the database (bacterial genomes from NCBI/RefSeq). d) Classification speed (in 10^3 reads per minute) using 1, 2, 4 and 8 threads. |
Registration is Open for the 2015 Bioinformatics Summer Workshops!
Now in its 8th year, the UC Davis Bioinformatics Training Program will be holding two week-long workshops this summer:
June 15-19, 2015: Using Command Line for Analysis of High Throughput Sequence Data
Sept 14-18, 2015: Using Galaxy for Analysis of High Throughput Sequence Data
These workshops will be held on the UC Davis campus and will run from 9am to 5pm on the dates indicated.
Details
Both workshops will cover modern high throughput sequencing technologies, applications, and ancillary topics, including:
Illumina HiSeq / MiSeq, and PacBio RS technologies
Read Quality Assessment & Improvement
Genome assembly
SNP, indel, and CNV discovery
RNA-Seq differential expression analysis
Experimental design
Hardware and software considerations
Cloud Computing
Each workshop will include a rich collection of lectures and hands-on sessions, covering both theory and tools. We will explore the basics of several high throughput sequencing technologies, focusing on Illumina and PacBio data for hands-on exercises. Participants will explore software and protocols, create and modify workflows, and diagnose/treat problematic data. Both workshops will utilize computing power of the Amazon Cloud (http://aws.amazon.com/),
In June, exercises will be performed using the Linux command line. Therefore, for this workshop, it is strongly recommended that participants should also have basic familiarity with the Linux/Unix (or Mac) command line.
In September, workshop exercises will be performed using the popular Galaxy platform (http://usegalaxy.org) which allows for powerful web-based data analyses. There are no prerequisites other than basic familiarity with genomic concepts.
Who Should Attend
Prior course participants have included faculty, post docs, grad students, staff, and industry researchers. Anyone with an interest in sequence analysis is welcome!
Registration Info
Attendance is limited to 36 participants per workshop in order to foster an effective learning environment and ensure sufficient one-on-one attention. Course tuition is $1,500 for academic or non-profit participants and $1,800 for other participants. Amazon has kindly provided grants of $100 per participant for Amazon Web Services accounts. This will allow you to perform analysis during and after the course using Amazon’s resources, without purchasing your own high performance computing servers!
To register, click on the links above or go to training.bioinformatics.ucdavis.edu/. All registration is “first-come, first-served”. There is no application process. We accept credit cards, as well as UC recharge accounts, for payment. Registration fees include light breakfast, lunch, and snacks, but do not include dinner, lodging or parking fees.
Questions
If you have any questions, please don’t hesitate to contact us:
See you this summer!
The UC Davis Bioinformatics Core Team
http://training.bioinformatics.ucdavis.edu
http://bioinformatics.ucdavis.edu/





