
Notes (I note – I am copying much of the text from the paper not rewriting it.)
What was sampled?
Ten surfaces (door handles into and out of the restroom, handles into and out of a restroom stall, faucet handles, soap dispenser, toilet seat, toilet flush handle, floor around the toilet and floor around the sink) in six male and six female restrooms evenly distributed across two buildings on the University of Colorado at Boulder campus were sampled on a single day in November 2010.
Surfaces where sampled using sterile, cotton-tipped swabs as described previously [14], [15]. As the 12 restrooms were nearly identical in design, we were able to swab the same area at each location between restrooms. In order to characterize tap water communities as a potential source of bacteria, 1 L of faucet water from six of the restrooms (each building having the same water source for each restroom sampled) was collected and filtered through 0.2 µm bottle top filters (Nalgene, Rochester, NY, USA).
How did they get DNA?
Genomic DNA was extracted from the swabs and filters using the MO BIO PowerSoil DNA isolation kit following the manufacturer’s protocol with the modifications of Fierer et al. [14].
How did they get sequence data?
A portion of the 16 S rRNA gene spanning the V1–V2 regions was amplified using the primer set (27 F/338R), PCR mixture conditions and thermal cycling conditions described in Fierer et al. [15]. PCR amplicons of triplicate reactions for each sample were pooled at approximately equal amounts and pyrosequenced at 454 Life Sciences (Branford, CT, USA) on their GS Junior system. A total of 337,333 high-quality partial 16 S rRNA gene sequences were obtained from 101 of the 120 surface samples collected, averaging approximately 3,340 sequences per sample (ranging from 513–6,771) (Table S1) in 4 GS Junior runs, with the best run containing 116,004 high-quality reads. An additional 16,416 sequences (ranging from 2161–5084 per sample) were generated for five of the six water samples collected for source tracking analysis. Each sample was amplified with a unique barcode to enable multiplexing in the GS Junior runs. The barcoded sequencing reads can be separated by data analysis software providing high confidence in assigning sequencing read to each sample. Sequence data generated as part of this study is available upon request by contacting the corresponding author.
How did they analyze the data?
All sequences generated for this study and previously published data sets used for source tracking (see below) were processed and sorted using the default parameters in QIIME [16]. Briefly, high-quality sequences (>200 bp in length, quality score >25, exact match to barcode and primer, and containing no ambiguous characters) were trimmed to 300 bp and clustered into operational taxonomic units (OTUs) at 97% sequence identity using UCLUST [17]. Representative sequences for each OTU were then aligned using PyNAST [18] against the Greengenes core set [19] and assigned taxonomy with the RDP-classifier [20]. Aligned sequences were used to generate a phylogenetic tree with FastTree [21] for both alpha- (phylogenetic diversity, PD)[22] and beta-diversity (unweighted UniFrac) [23] metrics. The unweighted UniFrac metric, which only accounts for the presence/absence of taxa and not abundance, was used to determine the phylogenetic similarity of the bacterial communities associated with the various restroom surfaces. The UniFrac distance matrix was imported into PRIMER v6 where principal coordinate analysis (PCoA) and analysis of similarity (ANOSIM) were conducted to statistically test the relationship between the various communities [24]. In order to eliminate potential biases introduced by sampling depth, all samples (including those used in source tracking) were rarified to 500 sequences per sample for taxonomic, alpha-diversity (PD), beta-diversity (UniFrac) and source tracking comparisons.
Sourcetracking
To determine the potential sources of bacteria on restroom surfaces and how the importance of different sources varied across the sampled locations, we used the newly developed SourceTracker software package [25]. The SourceTracker model assumes that each surface community is merely a mixture of communities deposited from other known or unknown source environments and, using a Bayesian approach, the model provides an estimate of the proportion of the surface community originating from each of the different sources. When a community contains a mixture of taxa that do not match any of the source environments, that portion of the community is assigned to an “unknown” source. Potential sources we examined included human skin (n = 194), mouth (n = 46), gut (feces) (n = 45) [26] and urine (n = 50), as well as soil (n = 88) [27] and faucet water (n = 5, see above). For skin communities, sequences collected from eight body habitats (palm, index finger, forearm, forehead, nose, hair, labia minora, glans penis) from seven to nine healthy adults on four occasions were used to determine the average community composition of human skin [26]. The mouth (tongue and cheek swabs), gut and urine communities were determined from the same individuals although the urine-associated communities were not published in the initial report of these data [26]. While urine is generally considered to be sterile, it does pick up bacteria associated with the urethra and genitals [28], [29]. The average soil community was determined from a broad diversity of soil types collected across North and South America [27].
—————————————————————
Notes on Sourcetracking
Abstract to paper:
Contamination is a critical issue in high-throughput metagenomic studies, yet progress toward a comprehensive solution has been limited. We present SourceTracker, a Bayesian approach to estimate the proportion of contaminants in a given community that come from possible source environments. We applied SourceTracker to microbial surveys from neonatal intensive care units (NICUs), offices and molecular biology laboratories, and provide a database of known contaminants for future testing.
Some lines from paper
We developed SourceTracker, a Bayesian approach to identifying sources and proportions of contamination in marker-gene and functional metagenomics studies. Our approach models contamination as a mixture of entire source communities into a sink community, where the mixing proportions are unknown.
SourceTracker’s distinguishing features are its direct estimation of source proportions and its Bayesian modeling of uncertainty about known and unknown source environments.
SourceTracker outperformed these methods (NAIVE BAYES AND RANDOM FORESTS) because it allows uncertainty in the source and sink distributions, and because it explicitly models a sink sample as a mixture of sources.
SourceTracker also assumes that an environment cannot be both a source and a sink, and we recommend research into bidirectional models.
Based on our results, simple analytical steps can be suggested for tracking sources and assessing contamination in newly acquired datasets. Although source-tracking estimates are limited by the comprehensiveness of the source environments used for training, large-scale projects such as the Earth Microbiome Project will dramatically expand the availability of such resources. SourceTracker is applicable not only to source tracking and forensic analysis in a wide variety of microbial community surveys (where did this biofilm come from?), but also to shotgun metagenomics and other population-genetics data. We made our implementation of SourceTracker available as an R package (http://sourcetracker.sf.net/), and we advocate automated tests of deposited data to screen samples that may be contaminated before deposition.
Who was there?
A total of 19 phyla were observed across all restroom surfaces with most sequences (≈92%) classified to one of four phyla: Actinobacteria,Bacteroidetes, Firmicutes or Proteobacteria (Figure 1A, Table S2). Previous cultivation-dependent and –independent studies have also frequently identified these as the dominant phyla in a variety of indoor environments [10]–[13]. Within these dominant phyla, taxa typically associated with human skin (e.g. Propionibacteriaceae,Corynebacteriaceae, Staphylococcaceae and Streptococcaceae) [30]were abundant on all surfaces (Figure 1A). The prevalence of skin bacteria on restroom surfaces is not surprising as most of the surfaces sampled come into direct contact with human skin, and previous studies have shown that skin associated bacteria are generally resilient and can survive on surfaces for extended periods of time [31], [32]. Many other human-associated taxa, including several lineages associated with the gut, mouth and urine, were observed on all surfaces (Figure 1A). Overall, these results demonstrate that, like other indoor environments that have been examined, the microbial communities associated with public restroom surfaces are predominantly composed of human-associated bacteria.
 |
|
Figure 1. Taxonomic composition of bacterial communities associated with public restroom surfaces.
(A) Average composition of bacterial communities associated with restroom surfaces and potential source environments. (B) Taxonomic differences were observed between some surfaces in male and female restrooms. Only the 19 most abundant taxa are shown. For a more detailed taxonomic breakdown by gender including some of the variation see Supplemental Table S2.
doi:10.1371/journal.pone.0028132.g001
|
Comparative analysis
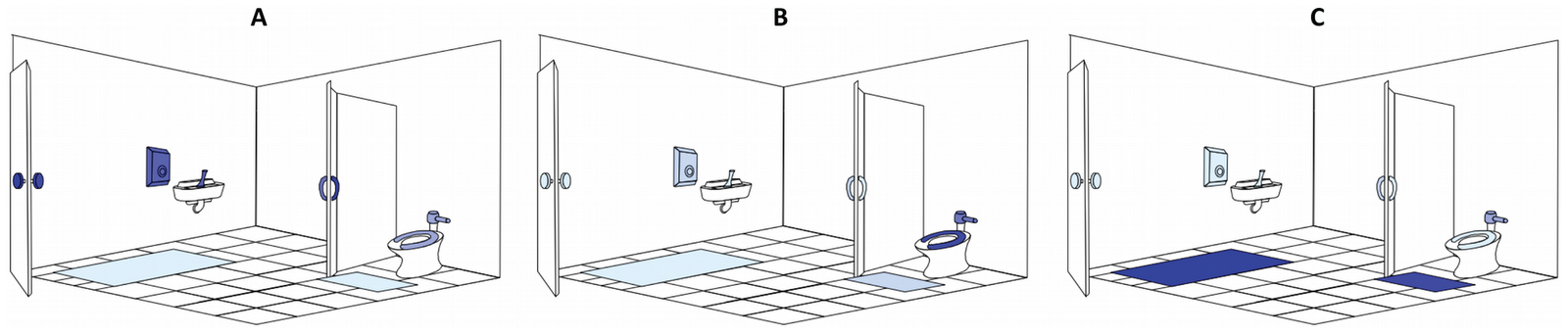
Comparisons of the bacterial communities on different restroom surfaces revealed that the communities clustered into three general categories: those communities found on toilet surfaces (the seat and flush handle), those communities on the restroom floor, and those communities found on surfaces routinely touched with hands (door in/out, stall in/out, faucet handles and soap dispenser) (Figure 2, Table 1). By examining the relative abundances of bacterial taxa across all of the restroom samples, we can identify taxa driving the overall community differences between these three general categories. Skin-associated bacteria dominate on those surfaces (the circles in Figure 2) that are routinely and exclusively (we hope) touched by hands and unlikely to come into direct contact with other body parts or fluids (Figure 3A). In contrast, toilet flush handles and seats (the asterisk-shaped symbols in Figure 2) were relatively enriched in Firmicutes (e.g.Clostridiales, Ruminococcaceae, Lachnospiraceae, etc.) andBacteroidetes (e.g. Prevotellaceae and Bacteroidaceae) (Figure 3B). These taxa are generally associated with the human gut [26],[33]–[35] suggesting fecal contamination of these surfaces. Fecal contamination could occur either via direct contact (with feces or unclean hands) or indirectly as a toilet is flushed and water splashes or is aerosolized [36]–[38]. From a public health perspective, the high number of gut-associated taxa throughout the restrooms is concerning because enteropathogenic bacteria could be dispersed in the same way as human commensals. Floor surfaces harbored many low abundance taxa (Table S2) and were the most diverse bacterial communities, with an average of 229 OTUs per sample versus most of the other sampled locations having less than 150 OTUs per sample on average (Table S1). The high diversity of floor communities is likely due to the frequency of contact with the bottom of shoes, which would track in a diversity of microorganisms from a variety of sources including soil, which is known to be a highly-diverse microbial habitat [27], [39]. Indeed, bacteria commonly associated with soil (e.g. Rhodobacteraceae, Rhizobiales, Microbacteriaceae and Nocardioidaceae) were, on average, more abundant on floor surfaces (Figure 3C, Table S2). Interestingly, some of the toilet flush handles harbored bacterial communities similar to those found on the floor (Figure 2, Figure 3C), suggesting that some users of these toilets may operate the handle with a foot (a practice well known to germaphobes and those who have had the misfortune of using restrooms that are less than sanitary).
 |
|
Figure 2. Relationship between bacterial communities associated with ten public restroom surfaces.
Communities were clustered using PCoA of the unweighted UniFrac distance matrix. Each point represents a single sample. Note that the floor (triangles) and toilet (asterisks) surfaces form clusters distinct from surfaces touched with hands.
doi:10.1371/journal.pone.0028132.g002
|
 |
|
Table 1. Results of pairwise comparisons for unweighted UniFrac distances of bacterial communities associated with various surfaces of public restrooms on the University of Colorado campus using the ANOSIM test in Primer v6.
doi:10.1371/journal.pone.0028132.t001
|
 |
|
Figure 3. Cartoon illustrations of the relative abundance of discriminating taxa on public restroom surfaces.
Light blue indicates low abundance while dark blue indicates high abundance of taxa. (A) Although skin-associated taxa (Propionibacteriaceae, Corynebacteriaceae,Staphylococcaceae and Streptococcaceae) were abundant on all surfaces, they were relatively more abundant on surfaces routinely touched with hands. (B) Gut-associated taxa (Clostridiales, Clostridiales group XI, Ruminococcaceae,Lachnospiraceae, Prevotellaceae and Bacteroidaceae) were most abundant on toilet surfaces. (C) Although soil-associated taxa (Rhodobacteraceae, Rhizobiales, Microbacteriaceae and Nocardioidaceae) were in low abundance on all restroom surfaces, they were relatively more abundant on the floor of the restrooms we surveyed. Figure not drawn to scale.
doi:10.1371/journal.pone.0028132.g003
|
Comparisons 2 (Gender)
While the overall community level comparisons between the communities found on the surfaces in male and female restrooms were not statistically significant (Table S3), there were gender-related differences in the relative abundances of specific taxa on some surfaces (Figure 1B, Table S2). Most notably, Lactobacillaceae were clearly more abundant on certain surfaces within female restrooms than male restrooms (Figure 1B). Some species of this family are the most common, and often most abundant, bacteria found in the vagina of healthy reproductive age women [40], [41] and are relatively less abundant in male urine [28], [29]. Our analysis of female urine samples collected as part of a previous study [26] (Figure 1A), found that Lactobacillaceae were dominant in urine, therefore implying that surfaces in the restrooms where Lactobacillaceae were observed were contaminated with urine. Other studies have demonstrated a similar phenomenon, with vagina-associated bacteria having also been observed in airplane restrooms [11] and a child day care facility [10]. As we found that Lactobacillaceae were most abundant on toilet surfaces and those touched by hands after using the toilet (with the exception of the stall in), they were likely dispersed manually after women used the toilet. Coupling these observations with those of the distribution of gut-associated bacteria indicate that routine use of toilets results in the dispersal of urine- and fecal-associated bacteria throughout the restroom. While these results are not unexpected, they do highlight the importance of hand-hygiene when using public restrooms since these surfaces could also be potential vehicles for the transmission of human pathogens. Unfortunately, previous studies have documented that college students (who are likely the most frequent users of the studied restrooms) are not always the most diligent of hand-washers [42], [43].
Source Tracking
Human sources:
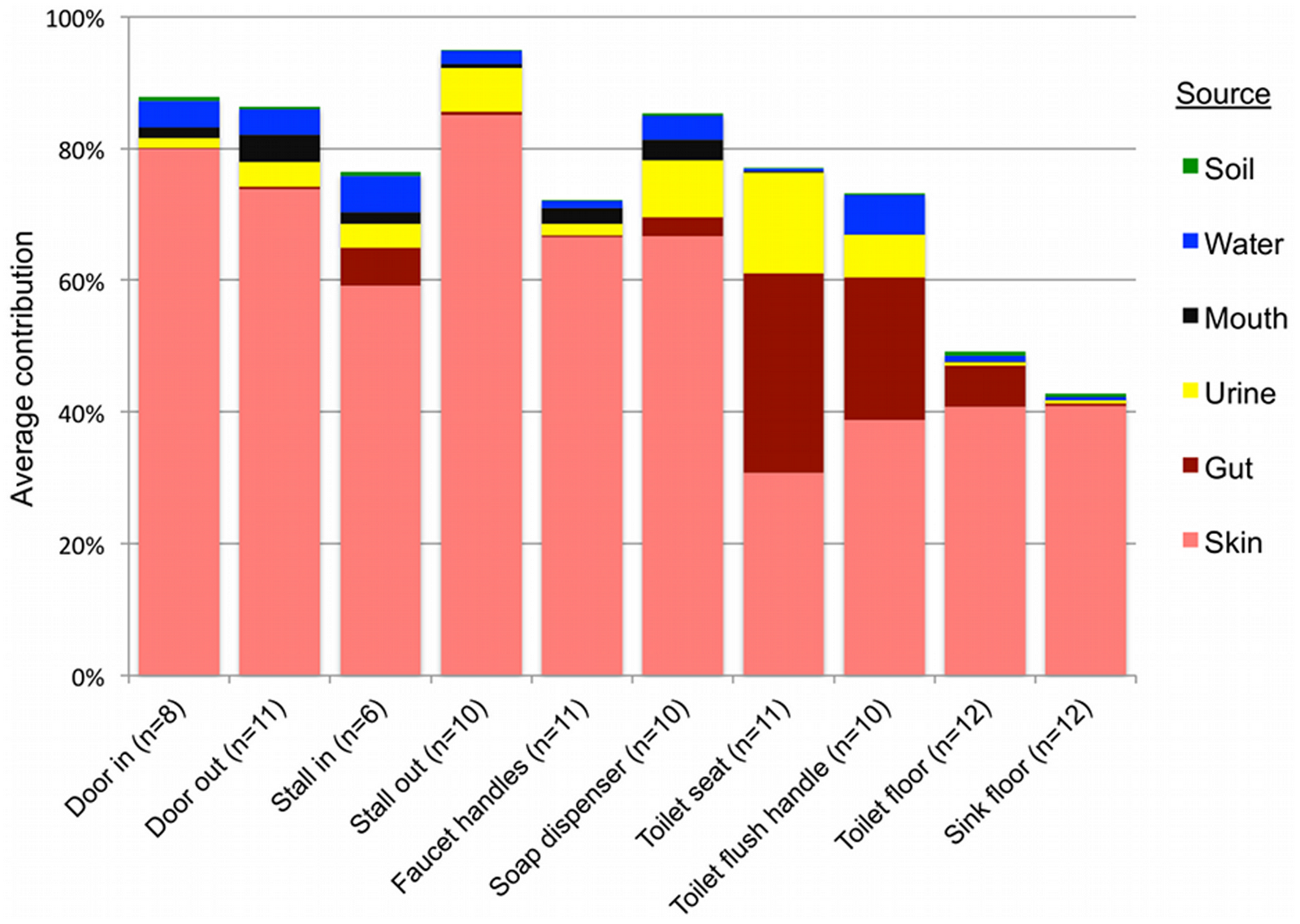
Results of SourceTracker analysis support the taxonomic patterns highlighted above, indicating that human skin was the primary source of bacteria on all public restroom surfaces examined, while the human gut was an important source on or around the toilet, and urine was an important source in women’s restrooms (Figure 4, Table S4).
Soil not an apparent source:
Contrary to expectations (see above), soil was not identified by the SourceTracker algorithm as being a major source of bacteria on any of the surfaces, including floors (Figure 4). Although the floor samples contained family-level taxa that are common in soil, the SourceTracker algorithm probably underestimates the relative importance of sources, like soils, that contain highly diverse bacterial communities with no dominant OTUs and minimal overlap between those OTUs in the sources and those found in the surface samples. As soils typically have large numbers of OTUs that are rare (i.e. represented by very few sequences) and the OTU overlap between different soil samples is very low [27], it is difficult to identify specific OTUs indicative of a soil source.
Other potential sources:
The other potential sources we examined, mouth and faucet water, made only minor bacterial contributions to restroom surface communities either because these potential source environments rarely come into contact with restroom surfaces (the mouth – we hope) or they harbor relatively low concentrations of bacteria (faucet water) (Figure 4). While we were able to identify the primary sources for most of the surfaces sampled, many other sources, such as ventilation systems or mops used by the custodial staff, could also be contributing to the restroom surface bacterial communities. More generally, the SourceTracker results demonstrate how direct comparison of bacterial communities from samples of various environment types to those gathered from other settings can be used to determine the relative contribution of that source across samples. Although many of the source-tracking results evident from the restroom surfaces sampled here are somewhat obvious, this may not always be the case in other environments or locations. We could use the same techniques to identify unexpected sources of bacteria from particular environments as was observed recently for outdoor air [44].
 |
|
Figure 4. Results of SourceTracker analysis showing the average contributions of different sources to the surface-associated bacterial communities in twelve public restrooms.
The “unknown” source is not shown but would bring the total of each sample up to 100%.
doi:10.1371/journal.pone.0028132.g004
|
While we have known for some time that human-associated bacteria can be readily cultivated from both domestic and public restroom surfaces, little was known about the overall composition of microbial communities associated with public restrooms or the degree to which microbes can be distributed throughout this environment by human activity. The results presented here demonstrate that human-associated bacteria dominate most public restroom surfaces and that distinct patterns of dispersal and community sources can be recognized for microbes associated with these surfaces. Although the methods used here did not provide the degree of phylogenetic resolution to directly identify likely pathogens, the prevalence of gut and skin-associated bacteria throughout the restrooms we surveyed is concerning since enteropathogens or pathogens commonly found on skin (e.g. Staphylococcus aureus) could readily be transmitted between individuals by the touching of restroom surfaces.










{kind=link}