
Here’s another entry in the “Story behind the paper series”. This one is from Lizzy Wilbanks, a co-advised PhD student in my lab (Twitter: @LizzyWilbanks)
A sulfurous symbiosis: Microscale sulfur cycling in the phototrophic pink berry consortia of the Sippewissett salt marsh
Here’s the story behind my recent publication (with many talented coauthors) on the pink berries, the marvelous, macroscopic microbial aggregates of the Sippewissett.
A bit of background:
The wild microbe rarely eats alone. The microbial world is a jungle far more exotic than those we can see (metabolically and phylogenetically, at least), one rife with fierce competition, intimate cooperation, and intricately inter-dependent food webs. Eavesdropping on the metabolic conversations of uncultured microbes, though, remains a major technical challenge. It requires tools to navigate the world from the microbe’s-eye view.
 |
| Your binoculars just aren’t gonna cut it… (image source ) |
Let’s get one thing straightened out:
 |
| (image source: my own, here, and here) |
My first encounter….
 |
| And people wonder why I think sulfide smells like beautiful summers and nostalgia? (image source: my own) |
 |
| (image source: my own) |
 |
| Me, awfully excited and really “diving-in” to the project. Can’t remember how many times TA Annie Rowe and others had to fish me out of the mud that summer! (image source: Melissa Cregger 🙂 |
Berries: an MBL Microbial Diversity legacy.
 |
| My obviously-not-to-scale cartoon of berry spearing with oxygen microsensors. |
 |
| Peering into the pink berries with a dissection microscope (real color!). Pink blobs are islands of purple sulfur bacterial cells. (image source: Verena Salman) |
 |
| The hypothesis! Purple sulfur bacteria in pink, sulfate reducing bacteria in green. (image source: my own, modified version of Figure 9 from our paper) |
Project launch: Team berry 2010
The first few weeks at the MBL course were bonanza of microbial excitement for me as a huge metabolism geek. My mornings were spent trying to drink from the fire hose of information in lecture, followed by afternoons of lab, then dinner, more lab, and finally trying to piece together the day’s ideas over beers.
 |
| “Drinking from a fire hose” – another gem from PhDComics |
Coming back from Dan Buckley and Victoria Orphan‘s lectures about the uses of stable isotopes in microbial ecology (reviewed here), I wondered if there was a way to use sulfur stable isotopes to track the cryptic sulfur cycle in the pink berries. Brainstorming with Victoria, we devised a plan to conduct incubations with the pink berries using isotopically heavy sulfate (34SO42-) as a stable isotope label. The purple sulfur bacteria in the berries had abundant intracellular sulfur reserves, which typically come exclusively from reduced forms of sulfur (e.g. sulfide). Our hope was that the sulfate reducing bacteria would reduce the heavy sulfate we added to heavy sulfide, which would then be oxidized by the purple sulfur bacterial and incorporated into their cells.
To track the flow of our isotopically labelled sulfur, we planned to image thin sections of the incubated berries using nanometer scale secondary ion mass spectrometry (nanoSIMS), an instrument commonly used by the Orphan lab for studying anaerobic methane oxidizing consortia.
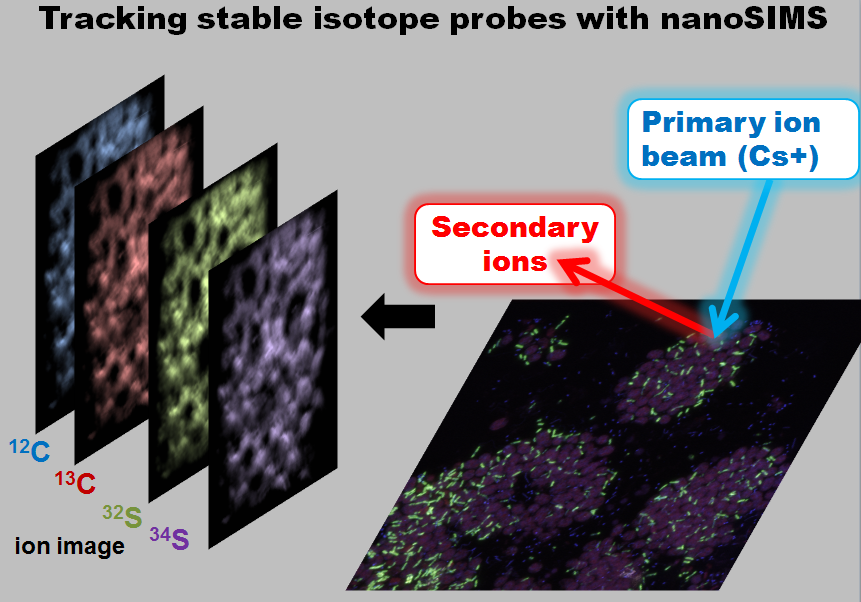 |
| Using the nanoSIMS to blast sections of pink berries with focused cesium beam (~50nm spot size) and generate spatial maps of isotopic and elemental abundance. (image source: my own). |
At that time, there was no precedent in the literature for using 34S-isotope labeling in this way (most stable isotope probing experiments focused on carbon or nitrogen compounds), but Victoria’s group was interested in exploring this area for studying other tightly coupled sulfur-cycling. The berries were an accessible testing ground. After a madcap two weeks of rush-orders, late nights, midnight berry slicing, and help from so many wonderful, patient TAs, our samples made a cross-country journey to the Orphan lab at Caltech where they, and thankfully the nanoSIMS, survived a minor earthquake.
 |
| The nanoSIMS beast in its subterranean lair @ the Caltech Microanalysis Center. (image source: my own) |
It was a wild ride during those final weeks, but just before the end, we got exciting results from Victoria’s nanoSIMS run that suggested our experiment had worked. The preliminary nanoSIMS data showed accumulation of our sulfur isotope label (enrichment in 34S compared to controls), and also found evidence for carbon fixation (13C enrichment from labeled bicarbonate additions).
Can’t stop, won’t stop… the side-project that ate my thesis.
After returning to Davis, passing my qualify exam and wrapping up prior projects, I was determined to get back to berries but wasn’t sure exactly how. Victoria suggested that she could include berries in a collaborative NSF proposal on the biogeochemistry of tightly coupled sulfur cycling consortia (along with David Fike, Greg Druschel and Jesse Dillon). When their funding came through, it held out the safety net I needed to work on berries full time. With approval from Victoria and my co-advisers at Davis, I jumped!
Returning as a TA to the MBL Microbial Diversity course in 2011, I had a chance to conduct follow up isotope experiments, and collaborate with course student and co-author Verena Salman on developing species-specific FISH probes to identify the spatial arrangements of the two berry symbiotic. Since then, I’ve followed up on our initial metagenomic sequencing to reconstruct near-complete genomes for the two berry symbionts, demonstrating the genetic potential for a complete sulfur cycle.
 |
| Figure 4 from our paper showing: the sulfate reducing species (green rods, 16S rRNA gene FISH probe) snuggled up with their metabolic partners, the purple sulfur bacteria (pink/purple cocci, autofluorescence), but not in the exopoylmer matrix with other cell types (blue, DNA stain: DAPI). |
In 2012, the final pieces of this project came together during a week of Sippewissett fieldwork with biogeochemistry collaborators David Fike, Greg Druschel, and their groups. With high resolution geochemistry equipment aboard our homemade raft, we were able to link our existing microbiological measurements with microscale geochemical signatures in the berries.
 |
| (image sources: my own) |
Conclusions:
FAQ:
- My naturalist’s answer is: because they’re the pink, charasmatic macrofauana of the microbial world. They’re nifty, and we don’t know what they do. But seriously…
- Microbial metabolism is the engine that drives the nutrient (biogeochemical) cycling that shapes the health of both our planet and our bodies.
- However, many key transformations in these cycles are carried out by microbial consortia over short spatiotemporal scales that elude detection by traditional analytical approaches.
- The berries provide a tractable, reproducible model microbial consortia for developing methods to eavesdrop on these otherwise cryptic metabolic conversations between the wild microbes.
- Understanding the biosignatures (e.g. sulfur isotopic fractionation) produced by microbial communities like the pink berries improves our ability to interpret the rock record and construct models of ecosystem function in both ancient and modern environments.
Thank you:
Through this project, I’ve had the privilege of working with truly amazing people and making life-long friends. The author list and acknowledgement are just the tip of the iceberg in terms of people who have contributed to this project in one way or another. You all know who you are; I feel so lucky to have gotten to know and work with you. THANK YOU!
This project was started as grass-roots style, curiosity-driven student research, and as such, the funding for it has been fairly eclectic. I want to take a moment to acknowledge those organizations that have supported this kind of research and made my work possible.
Funding to the MBL Microbial Diversity course from:
- Howard Hughes Medical Institute
- Gordon and Betty Moore Foundation (#2493)
- National Science Foundation (DEB-0917499)
- US Department of Energy (DE-FG02-10ER13361)
- NASA Astrobiology Institute (NAI)
Grants to collaborators Victoria Orphan and David Fike from:
- NSF (EAR-1124389 & EAR-1123391)
- Gordon and Betty Moore Foundation (#3306)
Grad-student grants and fellowships supporting my work at UC Davis from:
- National Science Foundation Graduate Research Fellowship
- UC Davis Dissertation Year Fellowship
- P.E.O. Scholar Award
- NAI/APS Lewis and Clark Fund in Astrobiology
- NSF Doctoral Dissertation Improvement Grant (DEB-1310168)
Full citation:
Wilbanks EG, Jaekel U, Salman V, Humphrey PT, Eisen JA, Faccioti MT, Buckley DH, Zinder SH, Druschel GK, Fike DA, Orphan VJ. (2014) “Microscale sulfur cycling in the phototrophic pink berry consortia of the Sippewissett Salt Marsh.” Environmental Microbiology, doi:10.1111/1462-2920.12388, http://dx.doi.org/10.1111/1462-2920.12388.








