
I am teaching a class this spring and as part of the class am having one lecture on “Phylogenetic trees and methods.” I would like to link to (and be able to mix and match material from) some review paper on this topic. So I am searching for something that is Open Access and preferably with a broad Creative Commons license. Anyone know of anything good?
Tag: Evolution

Phyloseminar.Org 3/29 Streaming talk by Jens Lagergren on Gene Family evolution
Just got his email from the organizer of Phyloseminar.Org:
On March 29th, phyloseminar.org will present Jens Lagergren speaking
on “Probabilistic analysis of gene families with respect with gene
duplication, gene loss, and lateral gene transfer.” Abstract below.
NOTE: the seminar will begin at 10h PST, which is three hours earlier
than the previous seminars.
This is 13h Eastern Standard Time, 19h Central European Time, and 6h
in Christchurch and Auckland!
Here’s the abstract:
Incongruences between gene trees and corresponding species trees are
common. Gene duplication, gene loss, and lateral gene transfer are
three types of evolutionary events that can cause such incongruences.
I will first describe a probabilistic process that contains standard
models of nucleotide substitutions (i.e., such that underly
probabilistic methods for phylogenetic tree reconstruction) as well as
gene duplication and gene loss. This process takes place in a given
species tree and can be used to reconstruct a gene tree for a gene
family of interest and simultaneously reconcile the gene tree with the
species tree. I will describe the algorithms available for this model
and also describe how they perform on biological data compared to
competing methods. Finally, I will describe an extension of this model
that also contains lateral gene transfer and show how it performs on
synthetic data.
Hope to see you there!
Share this:
Carnival of Evolution #20! is out and it’s got some good stuff …
Just a quick post here to suggest people check out the Carnival of Evolution (#20) being hosted at Skeptic Wonder (see Skeptic Wonder: Carnival of Evolution #20!).
It’s has some juicy evolution posts discussed and (perhaps) best of all has a “phylogenetic” tree based on the postings. I recommend everyone check it out …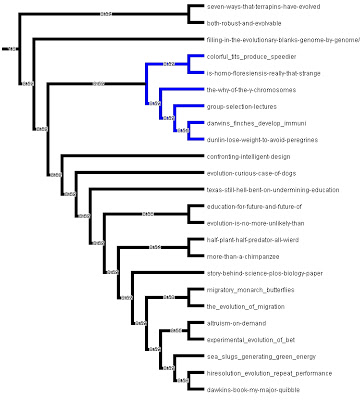
Share this:

Story behind the science: #PLoS Genetics "Evolutionary mirages" paper
![]()
So there is this cool new paper out in PLoS Genetics: Evolutionary Mirages: Selection on Binding Site Composition Creates the Illusion of Conserved Grammars in Drosophila Enhancers. and I have wanted to write about it for a week or so. You see, the paper is about something I have been interested in for most of my career – how the particular processes by which mutations occur can sometimes be biased (i.e., some types of mutations are more common than others) and that these biases can create highly ordered patterns in genomes and in turn that observation of these ordered patters can sometimes be misinterpreted as being the result of adaptation. Mistaken claims of adaptation in genomics are a favorite topic of mine – and let me to create (with tongue in cheek) a new omics word – Adaptationomics.
Anyway – so I really really like this paper. But there is a week bit of a problem in writing about it. You see, it is by my brother, Michael Eisen, a Prof. at UC Berkeley (and a student in his lab Richard Lusk). And, well, I don’t want to say anything wrong or stupid about the paper since, well, my brother will be pissed off. And so I have not written about it yet. But then I realized the best way to write about this one is to simply ask my brother for the “Story behind the science” for the paper, as I have been doing for some other recent papers.
If you want a summary of the paper, here it is in their own words:
Authors summary: Because mutation is a random process, most biologists assume that apparently non-random features of genome sequences must be the result of natural selection acting to create and preserve them. Where this is true, genome sequences provide a powerful means to infer aspects of molecular, cellular, and organismal biology from the signatures of selection they have left behind. However, recent analyses have shown that many aspects of genome structure and organization that have traditionally been attributed to selection can often arise from random processes. Several groups—including ours—studying the sequences that specify when and where genes should be produced have identified common, seemingly conserved, architectural features, based on which we have proposed new models for the activity of the complex molecular machines that regulate gene expression. However, in the work described here we simulate the evolution of these regulatory sequences and show that many of the features that we and others have identified can arise as a byproduct of random mutational processes and selection for other properties. This calls into question many conclusions of comparative genome analysis, and more generally highlights what Michael Lynch has called the “frailty of adaptive hypotheses” for the origins of complex genomic structures.
Conclusions: Lynch has eloquently argued that biologists are often too quick to assume that organismal and genomic complexity must arise from selection for complex structures and too slow to adopt non-adaptive hypotheses. Our results lend additional support to this view, and extend it to show that indirect and non-adaptive forces can not only produce structure, but also create an illusion that this structure is being conserved. We do not doubt that many aspects of transcriptional regulation constrain the location of transcription factor binding sites within enhancers. Indeed a large body of experimental evidence supports this notion, and we remain committed to identifying and characterizing these constraints. But if this process is to be fueled by comparative sequence analysis, as we believe it must be, it is essential that we give careful consideration to the neutral and indirect forces that we now know can produce evolutionary mirages of structure and function.
I must say I love the title lead in “Evolutionary mirages” which is another but much better way of saying “Adaptationism is a bad thing”.
Anyway, before I get in any more trouble, here are some words about the paper from the Senior Author, Michael Eisen, my brother. Questions by me (I know, not very creative ones – but they will have to do):
1. Why did you do this work?
This paper started out as a control. My lab is interested in understanding how the enhancers that control gene expression work – focusing on those that control early development in Drosophila. In 2008, we published a paper showing that when we put enhancers from a distantly related family of flies into Drosophila melanogaster embryos, they drive patterns of expression that are identical to the endogenous D. melanogaster enhancers, even though they have almost no conservation of primary DNA sequence. But since they have the same function, they must have something in common – and so we compared the configurations of transcription factor binding sites in orthologous enhancers across different evolutionary timescales looking for something they shared.
What we found is that binding sites in all of these enhancers occur in clusters. They are closer to each other than one would expect if they were scattered randomly in the ~1,000 bp of an enhancer. And, what’s more, sites that were close to each other were far more likely to be conserved. Surely, we thought, this could be no accident. So we proposed that enhancers are organized into compact clusters of sites for one or more factors – and that these “mini modules” are the primary unit of enhancer function.
But as we worked to extend these analyses to whole genomes, we sought a more rigorous, quantitative assessment, of just how improbably different levels of binding site clustering were. Like pretty much everyone in the field, we had used a null model in which binding sites were scattered randomly in an enhancer. But, I’ve been working with genomes long enough to know that nothing is ever truly random – and that all kinds of adaptive and non-adaptive processes create patterns in genome sequences that confound simple analyses. I wanted to come up with a null model for the distribution of sites within in an enhancer that was more realistic.
To do this I turned to my graduate student Rich Lusk, a card-carrying population geneticist trained at the University of Chicago. Rich was proud of his status as one of the few members of the lab who didn’t work on flies – but I convinced him to put aside the abstract models of binding site evolution in yeast and work on developing a real null model for our studies of enhancer evolution.
The idea was to simulate enhancers evolving without any constraint on the organization of transcription factor binding sites they contain, and to see what happens. But this did not mean letting enhancers evolve neutrally – their extreme functional conservation demonstrates that they are under fairly strong constraint. Since it is pretty clear that these enhancers are responding to the same transcription factors in all of these species, Rich’s simulations required that enhancers maintain their binding site composition – but placed no constraints on how the sites were organized relative to each other.
And what we found was striking. Even with no explicit selection on binding site organization – these evolved enhancers had lots of structure! Binding sites were clustered together, and, the closer together sites were, the more conserved they were — just like they were in real enhancers. In made us realize pretty quickly that the patterns we had latched onto – and which many other people were describing in different systems – might not be an evolutionary signature contraint on the organization of sites within in enhancers, but simply a byproduct of selection on binding site composition. If you want details, read the paper! But this has radically altered the way that we look at enhancer evolution.
2. How did you come up with the title.
Rich and I were writing the paper, and we had some really long, hideous, boring title. In writing the paper, the idea that things are not always what they appear to be was at the forefront of my mind. I was thinking about how desperate we and other people in the field were to figure out how enhancers work – it’s a vexing problem that has defied decades of work – and how we all hoped that evolutionary analysis was going to rescue us – and how quickly and eagerly we latched on to the first signs of a signal – and how that was just like a mirage you see in the desert….
3. Any interesting background?
(see 1)
4. When did the work start?
About a year ago. We had been thinking about this for a while, but only when Rich focused on it did things get rolling.
5. Why PLoS Genetics? Did PLoS Biology reject it?
PLoS Genetics was our first choice. PG has become the premier journal for evolutionary genetics – it routinely publishes the most interesting and important work in the field, and everyone reads it. While every paper I’ve sent there has been heavily scrutinized, the editorial process has been fair (though sometimes agonizingly slow….), and each review has been thoughtful and many (including in this case) helped to vastly improve the paper.
Lusk, R., & Eisen, M. (2010). Evolutionary Mirages: Selection on Binding Site Composition Creates the Illusion of Conserved Grammars in Drosophila Enhancers PLoS Genetics, 6 (1) DOI: 10.1371/journal.pgen.1000829
http://friendfeed.com/treeoflife/d5f1a668/story-behind-science-plos-genetics?embed=1
Share this:

Confronting Intelligent Design arguments directly in the scientific literature
![]()
A representative from Wiley publishing sent me a link to an interesting new paper. Entitled “Using Protistan Examples to Dispel the Myths of Intelligent Design” by Mark Farmer, from the University of Georgia and Andrea Habura, from the University at Albany, New York. It is from the Journal of Eukaryotic Microbiology and is based upon a presentation they gave at a workshop at a conference.
Basically, the article is a detailed discussion of how examples relating to microbial eukaryotes (I hate the term protist …) that are used by Intelligent Design advocates are, well, BS. And the article discusses the evidence that refutes the ID arguments.
One thing they discuss is the issue of the Cambrian Explosion. ID supporters, such as Stephen Meyer have made many arguments about they feel the diversification in the Cambrian is not explainable through evolutionary processes. Farmer and Habura refute this by pointing out that the diversity seen in microbial eukaryotes at the time of the Cambrian was immense and that what came out of the “explosion” was actually not that spectacular relative to what already existed in the microbial eukaryotes:
The extant diversity of the protists should therefore be seen as the “background radiation” of the eukaryotic Big Bang, with the Cambrian radiation of the metazoa being a subsequent event within a specific group.
They go on to discuss examples involving speciation, the fossil record, evolution of drug resistance in Plasmodium, and a few other things. In each case they discuss a claim by ID supporters and then discuss evidence for why this claim is not valid. Overall the paper is worth reading if you are involved in any discussions with ID supporters.
I note that when I finished the above writing, I went to look at Pubmed to find other examples of people taking on ID arguments in the literature with a focus on issues in microbes. Here are two other recent examples:
- Introduction: Protistan Biology, Horizontal Gene Transfer, and Common Descent Uncover Faulty Logic in Intelligent Design.
- Bacterial flagella and Type III secretion: case studies in the evolution of complexity.
Some discussion of this has now popped up on the web:
FARMER, M., & HABURA, A. (2010). Using Protistan Examples to Dispel the Myths of Intelligent Design Journal of Eukaryotic Microbiology, 57 (1), 3-10 DOI: 10.1111/j.1550-7408.2009.00460.x
Share this:

Story behind the science: #PLoS Biology paper on cichlid vision evolution
I am continuing on a new theme here in trying to get author feedback on recent PLOS publications. Today I write about a recent paper on PLoS Biology “The Eyes Have It: Regulatory and Structural Changes Both Underlie Cichlid Visual Pigment Diversity” by Christopher Hofmann, Kelly O’Quin, N. Justin Marshall, Thomas Cronin, Ole Seehausen and Karen L. Carleton
This paper discusses “how changes in gene regulation and coding sequence contribute to sensory diversification in two replicate radiations of cichlid fishes.” A good overview of the paper is in an accompanying article “Visual Tuning May Boost African Cichlid Diversity” by Robin Meadows:
“African cichlid fish form new species faster than any other vertebrates, with hundreds of species evolving within the last 2 million years in Lake Malawi and within the last 120,000 years in Lake Victoria. This rapid speciation makes cichlids good models for elucidating the genetic mechanisms behind biodiversity. Vision may play a key role in cichlid evolution, adapting them to forage for new foods or colonize new habitats. Vertebrate retinas have two groups of light-sensitive proteins called opsins: those in rod photoreceptors, which are sensitive to dim light, and those in cone photoreceptors, which are sensitive to color. Changes in the visual system could be due to differences either in the expression of opsin genes or in their DNA sequences. A Research Article in this issue of PLoS Biology by Christopher Hofmann and colleagues suggests that both mechanisms underlie changes in visual sensitivity in cichlids.”
For more on the science, see her summary and see the article itself. Additional information can be found in the press release from U. MD
But what I wanted to cover here was some of the story behind the science. So I emailed the authors some questions which they were kind enough to answer and I post the details here. There are some really interesting tidbits in these answers in my opinion, including how they dealt with merging two papers into one, and how difficult (but fun) it is to do this field work in Lake Malawi.
1. What led you to do the study reported in the paper?
From Karen Carlton:
This study was a long time in the making. We started studying the visual system of cichlids in the 1990’s. We learned quickly that there was a lot of variation in opsin expression within the Lake Malawi species. However, we had only examined a few species. In 2005, Tom Cronin and Justin Marshall (world experts on aquatic visual systems) agree to come to Lake Malawi with us and help examine a greater number of species. Justin brought his underwater spectrometer and characterized the light environment. Tom and I measured fish colors (that paper is under review) and I extracted retina for quantifying gene expression.
Because Lake Malawi and Lake Victoria both contain large cichlid radiations and had such different light environments, Ole Seehausen and I started working together in 2000 to compare visual systems in Malawi and Victoria. (Ole is the world expert on Lake Victoria cichlids, having helped discover the large rock dwelling species flock that escaped the devastation of the Nile perch). We concentrated on opsin sequences in our previous publications. However, we wanted to look at gene expression as well.
I was fortunate in 2006 to move to U Maryland where Chris Hofmann and Kelly O’Quin joined in our efforts. Chris took on the Victoria cichlid gene expression based on samples that Ole had collected. Kelly became our statistical wizard and analyzed the Malawi data we had gathered. (He has also been working on the visual system of Tanganyikan cichlids, which are the ancestors of the Malawi and Victoria flock. This work has recently been submitted).
From Kelly:
I see Karen gave you a nice review of how this paper was started. As she said, the work was started before I joined her lab. At that time, we were primarily concerned with moving into the new lab at UMCP, so no one was actively working on the data set. I initially analyzed the data to practice for a similar study of Tanganyikan cichlids. But, as I learned more about the power (and pitfalls) of the comparative analysis, I became more and more involved with the actual analysis and discussions, and after about 6 months Karen asked me to write up the paper for the Lake Malawi data set. At the same time Chris was working on a manuscript for the Victorian data. After seeing the overlap in the two papers — really the similarities and differences — Karen and Chris and I decided it would be useful to put the two together.
2. How did this group come together, with people from Australia, Switzerland and Maryland?
From Karen:
Vision science is a small international community that is wonderfully supportive. The cichlid community is also small and makes for excellent collaborations. This is what makes research great – combining expertise from such a diverse group of people. This enables us to think across many disciplines from physics to biology and integrate light measurements, ecology, molecular biology and genetics to try and understand what drives cichlid visual communication and determine how it plays a role in speciation.
From Christopher
I would add that both Europe and Australia have some top people in the field of visual ecology. Also, I don’t think we could have had a paper with such a broad scope without our collaborators. Once we all got together things just kept building and was very exciting.
3. A question for Kelly — how do you feel about the “joint contribution” statement. Do you think there needs to be a system to truly list two first authors or do you think this statement will suffice?
From Karen
I feel like I should chime in here. We originally had written two separate papers with Chris as lead author on the Victoria data and Kelly on the Malawi data. However, we all felt a combined paper could be more powerful. I asked Kelly and Chris to combine these papers, though that was a very difficult thing to ask, particularly in these times of first author is best. However, this paper is truly the joint effort of these two as well as the rest of the authors and would not be the paper that it is without everyone’s contributions and perspectives.
From Kelly
It is nice to be recognized for the work and effort given, and presumably this is accomplished in the ‘Author Contributions’ statement as well as the order in which authors are listed in. For this paper, Chris and I each authored manuscripts that Chris had to painstakingly combine. After a lot of debate over the meaning and limits of our comparative results, we each wrote a new drafts of the combined study that Karen then resolved into a single manuscript. Tom, Justin, and Ole provided lots of comments and additional text throughout this process as well. This truly was a collaborative effort, with plenty of contribution and compromise on everyone’s part. Although the order in which the author’s are listed cannot possibly communicate all of the nuances involved (though I am certainly happy with the order given), I hope we were able to addressed them with the ‘joint contribution’ statement you mention, as well as our ‘Author contributions’ statement (which lists just about every author under each category).
In short, I don’t think a simple change to the way that we list authors will ever capture all of the individual and combined efforts that go into a study. Instead, I think we need to change the way we read and interpret that list.
4. How did you end up choosing PLoS Biology as a place to submit the paper to? Were there any debates among the group about publishing there?
From Karen:
Online journals, such as PLoS Biology, give us a lot of flexibility to include all the supporting data without limiting the length of the paper.
From Kelly:
Since we had essentially two large studies here, the generous space and supplemental information limits allowed by PLoS made it a natural choice to publish in.
5. Do you have any good stories about the field work?
From Karen:
Field work in Malawi is never dull. Getting there is the first problem. It is a 24 hr plane ride if all goes well (which it never does) plus a 5 hr drive down to the lake, partly on Malawi dirt roads. Once you get there, however, the lake is a beautiful place. The diving is about the best in the world and it is wonderful to immerse yourself in your organism’s habitat. Underwater, it is wall to wall fish, with 50 or more species in a single location so it is perfect for observing and collecting a wide diversity of species.
The field station is run by the University of Malawi. It is right next to Chembe village and the people there are incredibly warm and friendly. The research station has electricity and cold running water. This is very high living for the village and makes for an interesting dichotomy. Several of the villagers are experts on cichlid fish, including Richard Zatha, and they dive with us. They can catch fish far faster than we can. There is considerable wildlife including the baboons which like to come into the house and steal bread off the table. We were fortunate in not having to deal with hippos or crocodiles on either of our recent trips.
It is quite expensive to take a group to Malawi. However, it is essential for everyone to see their organism in its natural habitat. It also takes a lot of preparation as well to get a group of scuba divers certified and ready to do this kind of field work.
I’m sure Ole has comparable stories for his work in Lake Victoria.
From Christopher:
To build on what Karen said. Going to Lake Malawi and actually diving with the fish is an incredible experience. When we work in our aquaculture facility we have maybe a handful of fish from a few different species in a single tank. In the field, once you drop below the surface it is an entirely different world. There are literally hundreds if not thousands of fish from many different species all doing their own thing. Some are eating algae, others plankton and even other fish. Many of these species are ones that are impossible to keep or breed in captivity, which makes the challenges of getting there worthwhile.
From Kelly:
Not really other than to say that it is a lot of hard work. But if you like SCUBA diving in remarkably clear water with beautiful, colorful fish, I can’t think of a better place to work than Lake Malawi.
6. Can you provide links to web sites of the authors and or other links of interest such as videos of the fish, twitter pages, etc?
- Our lab web site is: http://cichlid.umd.edu/cichlidlabs/kc/carletonlab.html
- Justin Marshall: http://ilc00f.facbacs.uq.edu.au/VTHRC/ecovis/
- Tom Cronin: http://www.umbc.edu/biosci/general/user/cronin
- Ole’s lab: http://www.eawag.ch/organisation/abteilungen/fishec/index_EN
7. Anything else you want to add:
From Christopher:
I would also add that its not easy to catch fish in Malawi. There is a definite art to scuba diving and handling a net. Having local cichlid experts was invaluable.
———————–
Cichlid picture by Christopher Hofmann doi:10.1371/journal.pbio.1000267.g001
![]()
Meadows, R. (2009). Visual Tuning May Boost African Cichlid Diversity PLoS Biology, 7 (12) DOI: 10.1371/journal.pbio.1000267
Hofmann, C., O’Quin, K., Marshall, N., Cronin, T., Seehausen, O., & Carleton, K. (2009). The Eyes Have It: Regulatory and Structural Changes Both Underlie Cichlid Visual Pigment Diversity PLoS Biology, 7 (12) DOI: 10.1371/journal.pbio.1000266
Share this:
Barcoding, taxonomy and citizen CSI
I just love the continued coverage of the story of the students from Trinity School in New York (a high school) who do investigative DNA barcoding projects. (There is a good new story about this on the LA Times blogs at:Think that sheep’s mik cheese comes from a sheep? DNA doesn’t lie | Booster Shots | Los Angeles Times)
In the most recent example, two students, Brenda Tan and Matt Cost, did some home barcoding in collaboration with people from the AMNH and Rockefeller University.
Among their findings:
- “an invasive species of insect in a box of grapefruit from Texas”
- “what could be a new species or subspecies of New York cockroach”
- multiple mislabelled food products including (quoted from the press release, I note)
- An expensive specialty “sheep’s milk” cheese made in fact from cow’s milk;
- “Venison” dog treats made of beef;
- “Sturgeon caviar” that was really Mississippi paddlefish;
- A delicacy called “dried shark,” which proved to be freshwater Nile perch from Africa;
- A label of “frozen yellow catfish” on walking catfish, an invasive species;
- “Dried olidus” (smelt) that proved to be Japanese anchovy, an unrelated fish;
- “Caribbean red snapper” that turned out to be Malabar blood snapper, a fish from Southeast Asia.
And what I find most interesting, is this built upon work of other students from Trinity Kate Stoeckle and Louisa Strauss who had done a restaurant based barcoding study last year.
This type of work is cool in so many ways. It gets students into science. It is an applied us of taxonomy (though I note, barcoding is not without controversy in the taxonomy community). It is a useful form of citizen science — and may eventually provide a way to keep dishonest sellers on their toes … Kudos to all involved in this
More on this story can be found at
Share this:
More coverage of the GEBA "Phylogeny Driven Genomic Encyclopedia"

Just a quick note here to post some links to additional stories about my new paper on “A phylogeny driven genomic encyclopedia of bacteria and archaea” which was published last week in Nature (with a Creative Commons license – which is rare in Nature but is what they use for genome sequencing papers).
Carl Zimmer has an article today in the New York Times “Scientists Start a Genomic Catalog of Earth’s Abundant Microbes” about the paper and the project. In the article he interviews me and Hans-Peter Klenk, who was a co-author and led the culturing part of the project. A few things to note about this:
- It is rare to have archaea mentioned in the New York Times.
- There is a tree that goes along with the article which is a modified version of the tree we had in our paper. I think theirs is very nice. Kudos to their artist
- There is a quote by Norm Pace generally supportive of the project
- The article mentions the JGI Adopt a Microbe program and even has a shout out to Malcolm Campbell at Davidson College and his recent PLoS One paper where they discuss results from a project where they took one of the genomes from our project and used it as part of a course on genome annotation/analysis.
For some of the story behind the paper see my recent blog post “Story Behind the Nature Paper on ‘A phylogeny driven genomic encyclopedia of bacteria & archaea’ #genomics #evolution“
Other discussions worth checking out
- John Timmer’s article for Ars Technica on “Presenting a genomic encyclopedia of bacteria (and archaea)”
- The Department of Energy is featuring the project as part of their “National Impact” Series” Scientists Launch the Genomic Encyclopedia of Bacteria and Archaea
- NYTimes Science Times discussion from Charlie Petit at the Knight Science Journalism tracker
Also see
- Archaea Make the Big Time from Genome Technology
- Woodland Daily Democrat (local paper): Encyclopedia of microbe genomes released
- Cory Golden at The Davis Enterprise wrote a nice story “Researchers urge new take on microbes” – not sure how long this stays online or how people access it though
- Microbe World has a bit on it
- Leonardo Martin has a really nice round up here
- The ScientificBlogging staff have written a bit about it here
- R&D mag Sr Editor Paul Livingstone has an interesting take on the story: Obsessive compulsive taxonomy
- Green Car Congress with mostly material from the press releases here.
- MyCor Web has a nice discussion of the paper
![]()
Wu, D., Hugenholtz, P., Mavromatis, K., Pukall, R., Dalin, E., Ivanova, N., Kunin, V., Goodwin, L., Wu, M., Tindall, B., Hooper, S., Pati, A., Lykidis, A., Spring, S., Anderson, I., D’haeseleer, P., Zemla, A., Singer, M., Lapidus, A., Nolan, M., Copeland, A., Han, C., Chen, F., Cheng, J., Lucas, S., Kerfeld, C., Lang, E., Gronow, S., Chain, P., Bruce, D., Rubin, E., Kyrpides, N., Klenk, H., & Eisen, J. (2009). A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea Nature, 462 (7276), 1056-1060 DOI: 10.1038/nature08656
Bakke, P., Carney, N., DeLoache, W., Gearing, M., Ingvorsen, K., Lotz, M., McNair, J., Penumetcha, P., Simpson, S., Voss, L., Win, M., Heyer, L., & Campbell, A. (2009). Evaluation of Three Automated Genome Annotations for Halorhabdus utahensis PLoS ONE, 4 (7) DOI: 10.1371/journal.pone.0006291
Share this:

Story behind the story for new #PLoSOne paper on Bayesian phylogenetics
![]()
There is an interesting new paper in PLoS One” Long-Branch Attraction Bias and Inconsistency in Bayesian Phylogenetics” by Brian Kolaczkowski and Joseph Thornton. The work focuses on methods for inferring phylogenetic history and in particular two types of statistical approaches: Likelihood and Bayesian. These methods are related to each other in that both attempt to use statistical models of evolution and then test different possible phylogenetic trees related taxa by how well certain data sets about those taxa map into the different possible trees. What they did in this new paper was test, with some simulations, and with some mathematical analyses. And somewhat surprisingly, they find that Bayesian methods, which have become more popular recently, appear to be more prone to errors than likelihood methods, when the data sets have multiple not closely related taxa with long branches. (Note if you want to learn more about phylogenetic methods, you can look at the online chapter (html format or PDF) from my Evolution Textbook, though I confess this needs a bit of revision, which I am working on now).
What they see in these cases is that the taxa with long branches group together, something known generally as “Long Branch Attraction” (LBA). Though there have been many previous studies of LBA, most have ended up showing that statistical methods are less prone to this problem than other phylogenetic methods, like distance and parsimony methods. What is surprising in this new work in that they find that Bayesian methods are highly prone to LBA – and much more so than likelihood methods.
Anyway, for more on this one could read the paper. But that I thought might be interesting is to ask the authors for more detail directly. I am hoping to do this more and more with PLoS papers in the future. I was inspired to do this, in fact, by one of the authors of this paper, Joe Thornton. He sent me an email with a link to the paper saying he thought I might be interested in it (true) and that he felt that it was his job in part for a PLoS One paper to make sure it got read by the right audience so he was hoping I might blog about it. And I said sure, but only if he gave me some of the “story behind the story”. So here it is below:
Why did you do these experiments?
Why did we do these experiments? A few years ago, we were studying the behavior of Bayesian posterior probabilities on clades — whether or not they accurately predict the probability that a clade is true, and what kinds of conditions might cause them to deviate from this ideal. We found that when the true tree was in the Felsenstein zone (two non-sister long branches separated by short branches), the long branches were often incorrectly grouped together with strong support. This was just a small part of a much larger paper that was published in MBE in 2008. The suggestion that Bayesian inference (BI) might be biased in favor of a false tree was surprising and intriguing, because we — like most people in the field — had assumed that BI would have the desirable statistical properties of ML (e.g., nearly unbiased inference and statistical consistency — convergence on the true tree with increasing support as the amount of data grows and the evolutionary model is correct, etc.). So we began doing experiments to rigorously explore the nature of the bias and its causes. When we found that BI was statistically inconsistent and the cause was integrating over branch lengths, we knew this result would be controversial, so we wanted to be sure the experiments were truly airtight. We supplemented our initial simulations with analyses of empirical data, with simulations under a wide variety of conditions using all types of priors, as well as mathematical and numerical analyses to clearly demonstrate the reasons for the bias. We also developed software that was identical to fully Bayesian MCMC except that it does not integrate over branch lengths; this method is not subject to the bias that BI displays, clearly demonstrating the cause of the bias.
Why did you send this to PLoS One?
Why did we submit to PLoS One? We think this paper has profound implications for phylogenetic practice and theory, and we want it to have a wide audience. Our experience with the review process in phylogenetic methods, unfortunately, is that many reviewers evaluate manuscripts based on whether or not the results confirm their world-view. This is a legacy of decades of internecine warfare in the field between the adherents of different methodological camps. We write papers in other fields, and while peer-review always has its ups and downs, our experience in phylogenetics is unusual in that solid papers are often rejected for philosophical reasons rather than for reasons of scientific validity and quality. We know this paper will be controversial, and we didn’t want it to be shot down in the review process for partisan reasons. PLoS One seemed like the perfect place to get the paper out and let the scientific community evaluate whether the experiments are convincing or not.
This is our first time publishing in PLoS One. I confess to being a little bit anxious that the paper will be lost in the great tide of papers published in the journal. We know our paper is very strong — I think it’s perhaps the most convincing and complete analysis of any problem I’ve ever published — so we’re confident that the work can have an impact, as long as the attention of readers in the field is drawn to it.
Where is the other author these days?
Bryan is now a postdoc in Andy Kern’s lab at Dartmouth.
Kolaczkowski, B., & Thornton, J. (2009). Long-Branch Attraction Bias and Inconsistency in Bayesian Phylogenetics PLoS ONE, 4 (12) DOI: 10.1371/journal.pone.0007891
Share this:

Nice Darwin Art at #UCDavis Evolution/Ecology Dept.



For more on this see The Face of Darwin where K. Garvey explains the history of the mural in more detail.


{kind=link}