Biomed Central web sites do such weird things when viewed in Safari …

 In fact, you could say in many ways we do very similar work, except he focuses on viruses. Not that we always agree mind you. I once gave a talk after him at a meeting and I changed my title to “Seeing the Forest and Missing the Trees” in a little dig at his not using phylogenetic methods and in his approach to metagenomic analysis. But I digress.
In fact, you could say in many ways we do very similar work, except he focuses on viruses. Not that we always agree mind you. I once gave a talk after him at a meeting and I changed my title to “Seeing the Forest and Missing the Trees” in a little dig at his not using phylogenetic methods and in his approach to metagenomic analysis. But I digress.
Alas, it is not freely available as it is in ISME but is not published under their “open” option. Am working on getting a link to an available PDF … will let everyone know.
Here is the abstract:
The Line Islands are calcium carbonate coral reef platforms located in iron-poor regions of the central Pacific. Natural terrestrial run-off of iron is non-existent and aerial deposition is extremely low. However, a number of ship groundings have occurred on these atolls. The reefs surrounding the shipwreck debris are characterized by high benthic cover of turf algae, macroalgae, cyanobacterial mats and corallimorphs, as well as particulate-laden, cloudy water. These sites also have very low coral and crustose coralline algal cover and are call black reefs because of the dark-colored benthic community and reduced clarity of the overlying water column. Here we use a combination of benthic surveys, chemistry, metagenomics and microcosms to investigate if and how shipwrecks initiate and maintain black reefs. Comparative surveys show that the live coral cover was reduced from 40 to 60% to 0.75 km2). The phase shift occurs rapidly; the Kingman black reef formed within 3 years of the ship grounding. Iron concentrations in algae tissue from the Millennium black reef site were six times higher than in algae collected from reference sites. Metagenomic sequencing of the Millennium Atoll black reef-associated microbial community was enriched in iron-associated virulence genes and known pathogens. Microcosm experiments showed that corals were killed by black reef rubble through microbial activity. Together these results demonstrate that shipwrecks and their associated iron pose significant threats to coral reefs in iron-limited regions.
Forest and others have recently been studying the Line Islands because they are relatively undisturbed reefs. Here are a short video about the work there (the work in general, not this specific study per se): http://oceantoday.noaa.gov/swf/flowplayer-latest.swf
Anyway, the new paper does something very different. It focuses on shipwrecks and the impact of these wrecks on reefs. This is of particular interest because as indicated in the abstract, the reefs are very low in iron. And many shipwrecks introduce massive amounts of iron. What they conclude in this new paper is that the iron from the shipwrecks leads to algal blooms, and lead to rapid killing of / damage to the pristine reefs.
For more on the paper there is an article in National Geographic Newswatch by Enric Sala worth checking out.
Forest also wrote me some information by email. He states:
Black reefs are associated with shipwrecks or other debris in this region of the world. These sites are interesting both from a conservation and scientific point of view. As a conservation issue, they are amazingly destructive. Kingman, one of the jewels of the USA coral reefs, has lost >1 km of the lagoon in less than 3 years. An old wreck on Fanning atoll has killed about 10% of their reef.
Visually, the black reefs are some of the eeriest places I’ve ever seen. The bottom is completely covered in different algae (including cyanobacterial mats), the water is filled with marine snow, and dark precipitate on the benthos (probably sulfur). We just published a paper in ISME where we have recreate the precipitate, cloudiness, and
coral death in microcosms by combining rubble from the black reefs, with corals and an iron addition. Addition of antibiotics blocks the coral death, precipitate, and marine snow, suggesting a microbial role.
The black reefs are probably caused by iron-enrichment from the wrecks and debris. We think black reefs are specific to non-emergent coral reefs, where iron is a limiting nutrient. Our current model is that iron stimulation of algae leads to increased microbial activity and coral death. In support of this, metagenomic analysis of the microbial community showed an enrichment of iron-related pathogenicity factors.
Forest also adds a plea to help in conservation of these reefs.
If you are interested in conservation, then please help us petition Congress to support removal of the wrecks and debris. Please contact Emily Douce at the Marine Conservation Biology Institute.
I encourage people to contact her.

Cool new paper from Joe Derisi’s lab: PLoS Biology: Chemical Rescue of Malaria Parasites Lacking an Apicoplast Defines Organelle Function in Blood-Stage Plasmodium falciparum. by Ellen Yeh and Joseph L. DeRisi. doi: 10.1371/journal.pbio.1001138
In it they use some experimental techniques to try and track down the elusive function of the apicoplast in Plasmodium falciparum, the causative agent of malaria. The apicoplast is an organelle that is evolutionarily derived from chloroplasts (and thus derived originally from cyanobacteria). Due to it’s cyanobacterial origins many have thought that it might serve as a good target for drugs to try and kill Plasmodium species because in theory such drugs if specific should not have significant detrimental effects on hosts like humans due to our lack of known important cyanobacterial associates.
Here is their abstract:
Plasmodium spp parasites harbor an unusual plastid organelle called the apicoplast. Due to its prokaryotic origin and essential function, the apicoplast is a key target for development of new anti-malarials. Over 500 proteins are predicted to localize to this organelle and several prokaryotic biochemical pathways have been annotated, yet the essential role of the apicoplast during human infection remains a mystery. Previous work showed that treatment with fosmidomycin, an inhibitor of non-mevalonate isoprenoid precursor biosynthesis in the apicoplast, inhibits the growth of blood-stage P. falciparum. Herein, we demonstrate that fosmidomycin inhibition can be chemically rescued by supplementation with isopentenyl pyrophosphate (IPP), the pathway product. Surprisingly, IPP supplementation also completely reverses death following treatment with antibiotics that cause loss of the apicoplast. We show that antibiotic-treated parasites rescued with IPP over multiple cycles specifically lose their apicoplast genome and fail to process or localize organelle proteins, rendering them functionally apicoplast-minus. Despite the loss of this essential organelle, these apicoplast-minus auxotrophs can be grown indefinitely in asexual blood stage culture but are entirely dependent on exogenous IPP for survival. These findings indicate that isoprenoid precursor biosynthesis is the only essential function of the apicoplast during blood-stage growth. Moreover, apicoplast-minus P. falciparum strains will be a powerful tool for further investigation of apicoplast biology as well as drug and vaccine development.
The author summary is a bit nicer in my opinion:
Malaria caused by Plasmodium spp parasites is a profound human health problem that has shaped our evolutionary past and continues to influence modern day with a disease burden that disproportionately affects the world’s poorest and youngest. New anti-malarials are desperately needed in the face of existing or emerging drug resistance to available therapies, while an effective vaccine remains elusive. A plastid organelle, the apicoplast, has been hailed as Plasmodium’s “Achilles’ heel” because it contains bacteria-derived pathways that have no counterpart in the human host and therefore may be ideal drug targets. However, more than a decade after its discovery, the essential functions of the apicoplast remain a mystery, and without a specific pathway or function to target, development of drugs against the apicoplast has been stymied. In this study, we use a simple chemical method to generate parasites that have lost their apicoplast, normally a deadly event, but which survive—“rescued” by the addition of an essential metabolite to the culture. This chemical rescue demonstrates that the apicoplast serves only a single essential function, namely isoprenoid precursor biosynthesis during blood-stage growth, validating this metabolic function as a viable drug target. Moreover, the apicoplast-minus Plasmodium strains generated in this study will be a powerful tool for identifying apicoplast-targeted drugs and as a potential vaccine strain with significant advantages over current vaccine technologies.
Also see their press release here.
Basically they are trying to use various experimental tricks to figure out which functions of the apicoplast are essential. Many theories have been proposed over the years as to what the apicoplast is doing. But few have gained significant evidence. This paper is an important contribution because it suggests that one pathway in particular is most functionally important: the isopentenyl pyrophosphate (IPP) synthesis pathway. See their model below:
| Figure 5. Model of apicoplast function. (Top) The essential function of the apicoplast is the production of isoprenoid precursors, IPP and DMAPP, which are exported into the cytoplasm and used to synthesize small molecule isoprenoids and prenylated proteins. Parasites that are unable to synthesize isoprenoid precursors either due to inhibition of the biosynthetic pathway by fosmidomycin or loss of the apicoplast following doxycycline inhibition can be chemically rescued by addition of exogenous IPP (red). The exogenous IPP enters the host cell through unknown membrane transporters and fulfills the missing biosynthetic function. (Bottom) Reaction scheme for MEP pathway biosynthesis of IPP and DMAPP with the enzymatic step inhibited by fosmidomycin indicated. |
Anyway – I have always been fascinated by apicoplasts because they are so weird. They reflect a strange evolutionary history of Apicomplexans in that this is a eukaryotic lineage that at some point brought into itself an entire photosynthetic algal cell as a symbiont. And for reasons still unknown (if there are reasons …) the chloroplast of the algal symbiont was retained while most of the rest of the symbiont was ditched. So that the resulting cells looked something like this:
 |
| From http://wiki.ericmajinglong.com/index.php?title=A_special_case:_The_apicomplexan_plastid |
Evolution is indeed very weird. And once it was discovered that the apicoplast was in fact derived from chloroplasts (this was discovered using molecular phylogenetics) (e.g., see http://www.sciencedirect.com/science/article/pii/016668519490149X) people have been wondering if it might make a good drug target. But people have also been wondering – what do Apicomplexans do with a chloroplast like organelle when they do not photosynthesize. So the Derisi paper is interesting both from a drug treatment point of view but also from an evolution point of view.
Anyway – here are some other links worth looking at:
Been working on revising my lab’s web site and was looking for some videos of talks I have given online to post there. And I discovered/rediscovered this video of an interview I did for Dr. Kiki’s Science Hour. Here it is:
NOTE – AT LEAST TEMPORARILY REMOVING THE VIDEO DUE TO MALWARE INFECTION OF TWIT.TV SITE
Now I know – this is over a year old. But I just watched the full video. Not so bad I think.
As many of you know, I like to talk. And talk. And talk. But I would like to say that as an interviewer, Dr. Kiki is pretty frigging awesome. Don’t know how she does it. But I am going to post this video on the new lab page and point people to it if they want to know what my lab does and what I am interested in.
But enough about me. I want to thank Dr. Kiki for this great interview by saying a little bit about her. Or, well, her work in science communication.
As some of you may know, I listen to podcasts of TWIS – This Week in Science frequently on my bike rides to work. And I really recommend anyone/everyone out there give it a whirl. It is sort of like Science Friday but it is a bit edgier, a bit funnier, a bit goofier, and a bit sciencier (is that a word?) Dr. Kiki and Justin on it are great and it is so good that I frequently sit outside my building listening to the end of a show if I take the short ride to work which is less than an hour. So if you like Science – you really should check out the TWIS web site and find some way to listen such as what I do by subscribing to their podcasts at iTunes.

And I guess now I will be checking out “Dr. Kiki’s Science Hour” more after rewatching this video. There are many many more shows at twit.tv/kiki. I have not checked out as many as TWIS shows but the ones I have watched are great.
And if you want to follow her more directly check out her Blog: The Bird’s Brain, or her twitter feed (@drkiki) or her Google+ feed.
Very proud that she is a UC Davis alum … and just want to say thanks to her for giving me a video I can share with others that says more about me and my lab than almost anything I have written.

Last year, Rebecca Skloot came to Davis to talk about her book “The Immortal Life of Henrietta Lacks“. Note – if you have not read the book – what f*$ing rock have you been hiding under? It is in my opinion the best non fiction book I have ever read. Seriously. Not the best science book. The best non fiction book of any kind. And I am not alone in this feeling as it has won a bazillion positive reviews and awards. In summary – it tells three stories – the story of the isolation of HeLa cells, the story of the woman from whom those cells came, and the story of Skloot learning the other two stories.
Anyway – I somehow managed to get her to come to UC Davis to give a talk last year just as the book was going viral. In preparation for Skloot’s visit I decided to do some sort of “open access” schtick and looked into how many papers about HeLa cells were in Pubmed Central. Pubmed Central is a database of papers for which the full text is available at no charge. After this mini-research and after interacting with Rebecca over the last year and seeing the well deserved recognition of her book, I have been a bit fascinated about how much of the literature surrounding studies of HeLa cells is openly and/or freely available.
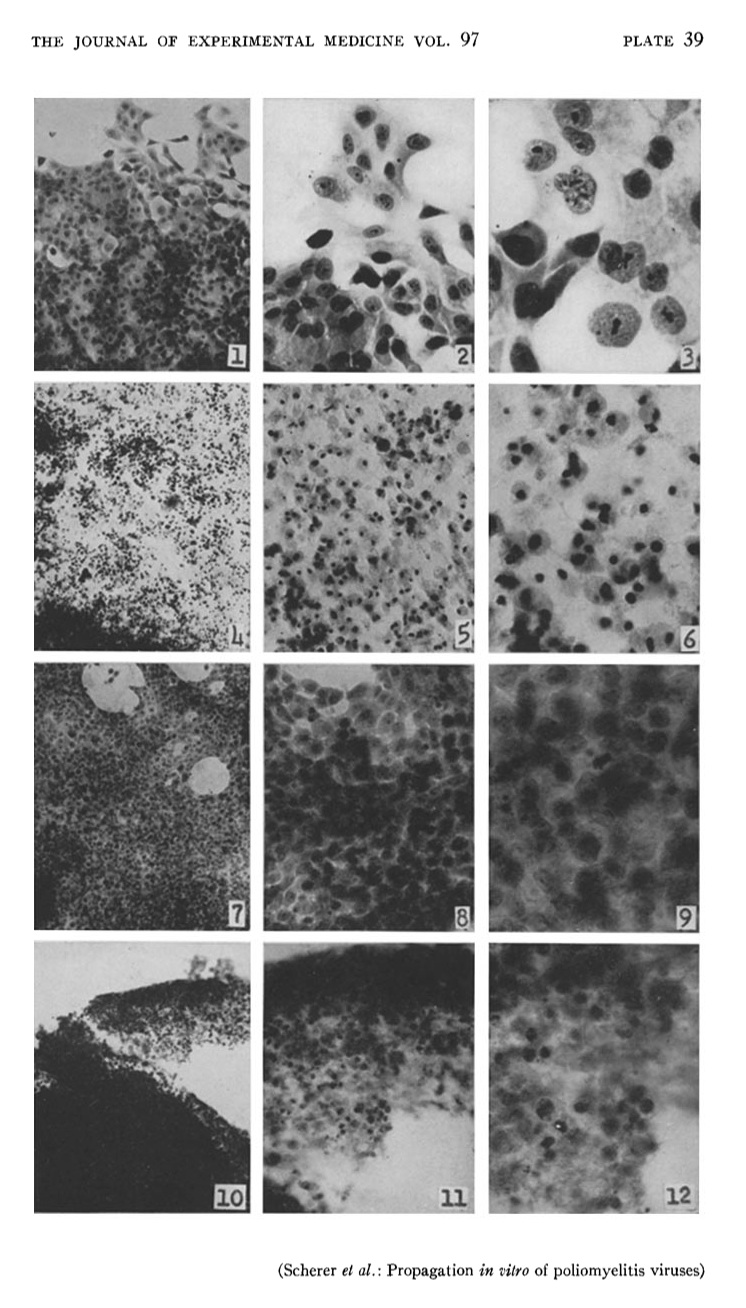
So today I decided to see what was the earliest HeLa paper that was freely available on PubMed Central. And I managed to find a good one: Studies on the propagation in vitro of poliomyelitis viruses. IV. viral multiplication in a stable strain of human malignant epithelial cells (strain hela) derived from an epidermoid carcinoma of the cervix. William F. Scherer, Jerome T. Syverton, and George O. Gey. J Exp Med. 1953 May 1; 97(5): 695–710.
I believe this was the first full paper published discussing HeLa cells. Nice short title by the way. Anyway – good to see it in Pubmed Central.
I note – I could not find in Pubmed Central the meeting abstract about HeLa cells which was published in 1952 but I did find it from Cancer Research’s online archive here. I have copied the abstract below, for you HeLa history buffs out there:
TISSUE CULTURE STUDIES OF THE PRO-
LIFERATIVE CAPACITY OF CERVICAL
CARCINOMA AND NORMAL EPITHE-
LIUM. George O. Gey,Ward D. Coffman*
and Mary T. Kubicek *(Departments of
Surgery and Gynecology, Johns Hopkins
Hospital and University, Baltimore 5, Md.This is a report of an evaluation in vitro of the
growth potential of normal, early intra-epithelial,
and invasive carcinoma from a series of cases of
cervical carcinoma. Comparable cytological and
tissue culture studies were actually carried out on
selected biopsies of normal and neoplastic areas of
the same cervix. Thus far, only one strain of epi-
dermoid carcinoma has been established and
grown in continuous roller tube cultures for al-
most a year. It grows well in a composite medium
of chicken plasma, bovine embryo extract, and
human placenta! cord serum. The autologous nor-
mal prototype is most difficult to maintain under
comparable cultural conditions. Most of the tissue
from other cases showed rapid keratinization of
the cells grown in cultures whether from normal
or neoplastic areas. Some of the hormonal aspects
of the problem will be discussed.
Pubmed Central is a rich resource not just for accessing scientific papers but for learning about science history too. It is a good thing that articles in Pubmed Central are available at no charge and here’s hoping that sometime soon that past and present and future science papers will be more readily available to all.
UPDATE ———-
John Hogenesch from U. Penn made a nice figure of relevance and agreed to let me post it:
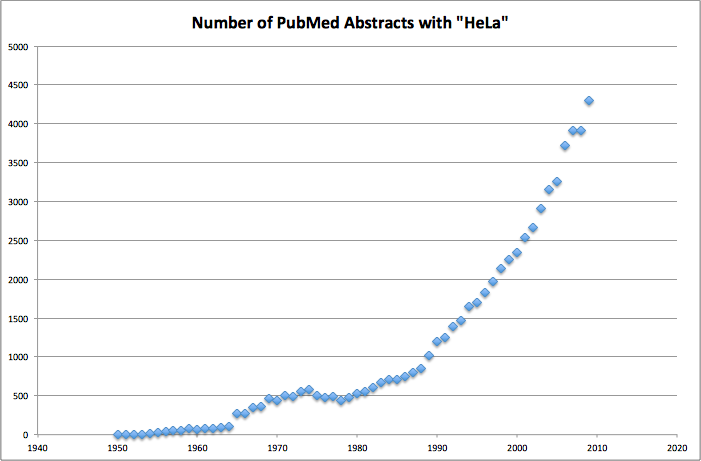
And he comments “You can see several things, Nixon’s “war on cancer” in the early 70s and the dawn of the cell/molecular biology age in the 80s and expansion in the 90s.”
Thanks John …
There is a new paper out: Phylogenetic-based propagation of functional annotations within the Gene Ontology consortium in Briefings in Bioinformatics.
The paper is interesting and presents a new general approach to using phylogeny for functional prediction of uncharacterized genes. I am interested in this for many reasons including that I was one of, if not the first to lay this out as a concept. In a series of papers from 1995-1998 I outlined how phylogenetic analysis could be used to aid in functional prediction for all the genes that were starting to be sequenced in genome projects without any associated functional studies (at the time, I referred to all these ESTs and other sequences as an “onslaught” – little did I know what was to come).
My first paper on this topic was in 1995: Evolution of the SNF2 family of proteins: subfamilies with distinct sequences and functions. The abstract is below:
The SNF2 family of proteins includes representatives from a variety of species with roles in cellular processes such as transcriptional regulation (e.g. MOT1, SNF2 and BRM), maintenance of chromosome stability during mitosis (e.g. lodestar) and various aspects of processing of DNA damage, including nucleotide excision repair (e.g. RAD16 and ERCC6), recombinational pathways (e.g. RAD54) and post-replication daughter strand gap repair (e.g. RAD5). This family also includes many proteins with no known function. To better characterize this family of proteins we have used molecular phylogenetic techniques to infer evolutionary relationships among the family members. We have divided the SNF2 family into multiple subfamilies, each of which represents what we propose to be a functionally and evolutionarily distinct group. We have then used the subfamily structure to predict the functions of some of the uncharacterized proteins in the SNF2 family. We discuss possible implications of this evolutionary analysis on the general properties and evolution of the SNF2 family.

I note – I am annoyed that when I went to the Nucleic Acids Research site for my paper I discovered for some bizarre reason they are now trying to charge for access to it even though it is in Pubmed Central and used to be freely available on the NAR site. WTF? Is this just an IT issue like the #OpenGate complaints I made for a while about Nature Genome papers.
Anyway – in that paper in 1995 I basically showed that at least for this family, phylogenetic analysis could be used as a tool in making functional predictions by allowing one to better identify orthology relationships and subfamilies within the SNF2 superfamily. This was novel I think maybe a little bit but others at the time were also looking into using various analyses to identify orthology relationships across genomes.
Shortly thereafter I started working on the concept that one could used the phylogenetic tree more explicitly in making functional predictions and eventually I laid out the concept of treating function as a character states and doing character state reconstruction using a gene tree to then infer functions for uncharacterized genes. I called this approach “phylogenomics” in a paper in 1997 in Nature Medicine (the editor asked us to give it a name … and thus my own contribution to the omics word game began). Alas somehow the title of our paper became “Gatrogenomic delights” a movable feast” since we were writing about the E. coli and H. pylori genomes, so I added yet another omics term at the same time. In the paper I showed how phylogenetic analysis of the MutS family of proteins could help in interpreting one of the findings in the H. pylori genome paper:
 |
In this paper we showed why blast searches were not ideal for inferring relationships among sequences (because blast measures similarity NOT evolutionary history per se). A bit annoyed still that other papers then sort of claimed they were the first to show blast was not ideal for inferring evolutionary relatedness, but whatever. This still did not fully describe the phylogeny driven approach that I was working on so I then wrote up an outline of this approach for a paper in Genome Research: Phylogenomics: Improving Functional Prediction for Uncharacterized Genes by Evolutionary Analysis. This paper really laid out the idea in more detail:
 |
||
It also gave detailed examples of how similarity searches could be misleading and how phylogenetic analysis should in principle be better.

I note – I am very very proud of this paper. But it did not do a lot of things. Really it was about laying out a concept of using tools from phylogenetics in functional prediction. But it did not provide software for example. I later developed some of my own scripts for doing this when I was at TIGR but really the software for phylogeny driven functional predictions would come later from others like Kimmen Sjolander, Sean Eddy, and Steven Brenner. Each method laid out in these tools and in other papers had its own flavors and I continued to explore various approaches and applications to phylogeny driven functional prediction. Examples of my subsequent work are listed below (with links to the Mendeley pages for these papers):
Plus we (at TIGR) used phylogenetic analysis as a tool in annotation of many many genomes as well as metagenomes.
Anyway, enough of history for a bit. What is interesting about this new paper is that they take a slightly different approach to phylogeny driven functional prediction in that they make use of Gene Ontology functional annotations as their key parameter to trace on evolutionary trees. They lay out the differences in their method quite well in the introduction:
Our general approach is similar to the ‘phylogenomic’ method proposed by Eisen [6] and further developed into a probabilistic form by Engelhardt et al. [7], but with important differences. Eisen proposed a conceptual approach for predicting protein function using a phylogenetic tree together with available experimental knowledge of proteins. The original approach relied on manual curation to identify gene duplication events and to find and assimilate the literature for characterized members of the family. Engelhardt et al. used automated reconciliation with the species tree [8] to identify gene duplication events, and experimental GO terms (MF only) to capture the experimental literature. Using this information, they defined a probabilistic model of evolution of MF involving transitions between different molecular functions.
From these previous studies, we adopt the basic approach of function evolution through a phylogenetic tree and the use of GO annotations to represent function. However, unlike these other phylogenomic methods, we represent the evolution in terms of discrete gain and loss events. In Eisen’s original model, an annotation does not necessarily represent a gain of function (it could have been inherited from an earlier ancestor), and losses are not explicitly annotated. The transition-based model of Engelhardt et al. assumes replacement of one function by another (gain of one function coupled to the loss of another), and does not capture uncoupled events, which is particularly important for BP annotations and cases where a protein has multiple molecular functions (see examples below). In addition, we make no a priori assumptions about conservation of function within versus between orthologous groups, or about the relationship between evolutionary distance and functional conservation (as the distance may not necessarily reflect every given function). While, as described below, gene duplication events and relatively long tree branches are important clues for curators to locate functional divergence (gain and/or loss), in our paradigm an ancestral function can be inherited by both descendants following a duplication (resulting in paralogs with the same function) or gained/lost by one descendant following a speciation event (resulting in orthologs with different functions). Evolution of each function is evaluated on a case-by-case basis, using many different sources of information about a given protein family
I note – Paul Thomas, one of the authors here has also been developing phylogeny driven functional prediction methods for many years and has done some cool things previously. This new approach seems novel and useful and their paper is worth looking at. I like too that they focus on MutS homologs for some of their examples:


Anyway – their paper is worth a read and some of their software tools may be of use including PAINT: http://sourceforge.net/projects/pantherdb/ and http://pantree.org
Good to see continuous developments in phylogeny driven functional predictions. If you want to learn more – check out the Mendeley Group I have created:
http://www.mendeley.com/groups/1190191/_/widget/29/5/
And please contribute to it. Below are some previous posts of mine of possible interest:
Recently a paper by Matt Hahn was published in PLoS Computational Biology entitled “Testing the ortholog conjecture with comparative functional genomic data from mammals.” The paper created a bit of a stir as some aspects of it call into question some of the standard assumptions made in comparative genomic analysis.
I alas do not have time to go into all the details but fortunately others have tackled this and I am posting some links here:
http://friendfeed.com/erickmatsen/f90bd2c6/emergentnexus-i-think-what-you-were-talking?embed=1
Will try to post my own comments soon. I note – I am skeptical of their conclusions but still going through the paper to understand everything before commenting in more detail.
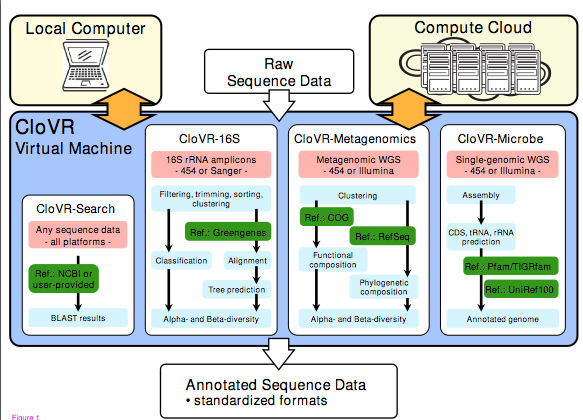
Not only is Clovr available openly and freely but they even have a Clovr blog: http://clovr.org/category/blog/ … though it does not seem to be heavily used. Kudos to this team for producing and releasing this software for others to use. And kudos to NSF, USDA and NIH for funding its development — I have a feeling many people will use it.
![]()
| Figure 2. Phylogenetic tree linking metagenomic sequences from 31 gene families along an oceanic depth gradient at the HOT ALOHA site |
I am a co-author on a new paper that came out in PLoS One yesterday. The paper is PLoS ONE: The Phylogenetic Diversity of Metagenomes and the full citation is Kembel SW, Eisen JA, Pollard KS, Green JL (2011) The Phylogenetic Diversity of Metagenomes. PLoS ONE 6(8): e23214. doi:10.1371/journal.pone.0023214.
The first author is Steven Kembel, a brilliant post doc at the University of Oregon. You can follow him on twitter here. This paper is a product of the “iSEEM” “integrating statistical, ecological and evolutionary approaches to metagenomics” collaboration between my lab and the labs of Jessica Green at U. Oregon and Katie Pollard at UCSF. For more on iSEEM see http://iseem.org. iSEEM was supported by the Gordon and Betty Moore Foundation.
Anyway – the paper focuses on developing and using a new method for assessing the phylogenetic diversity of microbes via in samples via analysis of metagenomic data. Phylogenetic diversity (aka PD) is measured by building evolutionary trees and summing up the total length of branches in such trees. It is an important diversity metric and is complementary to metrics such as “species richness” which is a measure of the number of species in a sample. When one counts species in a sample, one ends up ignoring the evolutionary distances between species and thus one may get an incomplete picture of the diversity of organisms in a sample simply by counting species. For example, a sample that contains 500 different species in the genus Escherichia would have the same “richness” as a sample that contained one representative of each of 500 different Orders of bacteria. For many purposes it is useful to know whether one has a phylogenetically diverse sample or not. (And of course, if one just focuses on species richness it is also important to not simply ignore some set of organisms in the samples as has sort of been done in a recent paper estimating the total species richness on the planet). But that is not the point here – the point here is that counting species, even if done correctly, can give an incomplete picture of the diversity of organisms in sample.
For many years researchers have been attempting to measure phylogenetic diversity of various organisms in various samples. And to do this one needs an evolutionary tree of the organisms in order to then measure branch length in the tree. There is actually a relatively rich history of researchers attempting to look at PD in studies of microbes – especially in cases where one has access to a rRNA tree for the organisms / samples in question. Examples of past work on this include:
I note, as an aside, I have created a Mendeley group focusing on phylogenetic analysis of metagenomes and have added a diversity of papers to the collection:
http://www.mendeley.com/groups/1152921/_/widget/29/2/


We want to build trees from these alignments with the hope of using them to learn lots of cool things about the evolution of the fragments and the species from which they come. I can provide more information but really the key part for the phylogenetics here is the nature of the alignment.
In the past, I have decided to constrain my analyses to NOT deal with this type of alignments. I have either analyzed each fragment on its own or we have built a multiple alignment but only inlcuded fragments that cover more than 3/4 of the full length sequence and thus the matrix is much more filled out. Such an alignment would look like this
But we really want to include the smaller fragments in our analysis. And we are just not certain how to best do this. We know LOTs of people out there think of similar problems in terms of sparse matrices, supermatrices, supertrees, EST data, etc. And we have ideas about how to do this and are asking around by email some phylogenetics gurus we know. But I thought it might be fun to have the discussion on a blog rather than by email.
So again, how might one best build phylogenetic trees from data that looks like this?
And from these trees we want to place each fragment relative to (1) the full length sequences and (2) to each other if possible. We also, of course, want branch lengths to reflect some sort of amount of evolution and thus do not just want a cladogram.
| Figure 1. Conceptual overview of approach to infer phylogenetic relationships among sequences from metagenomic data sets. |
| Phylogenetic tree linking metagenomic sequences from 31 gene families along an oceanic depth gradient at the HOT ALOHA site |
And from that he could calculate PD for metagenomic samples. We then used the PD calculations to comparate and contrast PD with other information in particular from the HOT ALOHA metagenomic data set of Ed Delong, Steve Karl and others.
| Figure 3. Taxonomic diversity and standardized phylogenetic diversity versus depth in environmental samples along an oceanic depth gradient at the HOT ALOHA site. |
For more detail on what we did from there on – read the paper. It is open access so all can see it / download it / play with it / whatever. But rather than blather on and on as usual I thought I would email Steve some questions and then post his answers. These are below:
This work got started as a collaboration between the Eisen, Green, and Pollard labs as part of the iSEEM project (“Integrating Statistical Evolutionary & Ecological Approaches to Metagenomics”), which was funded by the Moore Foundation to figure out ways to address ecological and evolutionary questions using metagenomic data. I had a background in using phylogenetic and evolutionary information to understand ecological communities, and one of the things I wanted to do at iSEEM was to try to think about ways that we could apply methods from ecophylogenetics or phylogenetic community ecology to metagenomic data sets. In conversations among the co-authors, we realized that if we could build phylogenetic hypotheses for organisms based on metagenomic data, we could apply a huge body of ecological and evolutionary theory and use these data sets to improve our understanding of microbial communities and their dynamics.
2. How did you end up working on microbes with your background in larger organisms?
The transition from working on macro-organisms to working on microbes actually wasn’t that big of a leap, since my research has generally been question driven rather than study-system or study-organism driven. My previous research involved using phylogenetic information to better understand community assembly in plants and animals. The increasing availability of phylogenetic information for entire communities of plants and animals drove the development of the field of ‘ecophylogenetics’, and it always seeemed to me that microbes would be the ideal system for this type of approach due to the greater availability of sequence data and phylogenetic information for microbes. Also, the development of high-throughput sequencing methods meant that the size of microbial community data sets would quickly become really, really large… the prospect of working on data sets with hundreds of millions of observations was really exciting. As my first postdoc was wrapping up, I collaborated on a study looking at phylogenetic diversity of the rhizobacterial symbionts of plant roots that got me interested in microbial ecology. Right around that time I came across the opportunity to work on the iSEEM project, so it seemed like the perfect opportunity to try a new study system.
Having studied the community ecology of both micro- and macro-organisms, I find it interesting that the fields of microbial and non-microbial phylogenetic community ecology have been fairly insulated from one another until recently. For example, the two fields independently developed phylogenetic approaches to community ecology, each field having its own set of favored statistical methods and software packages, with almost no cross-citation, despite addressing very similar questions. In microbiology the emphasis on phylogenetic diversity measures seems to have been driven by the empirical difficulty of defining microbial ‘species’ and other taxonomic units that macro-organismal ecologists are comfortable with, as well as the availability of phylogenetic and sequence data for microbes. Conversely, for macroorganisms the field of ecophylogenetics was driven by a desire to apply a large body of theory on the links between ecological and evolutionary dynamics to empirical data sets, but was relatively data poor in terms of phylogenetic information about individual species.
3. What was the biggest challenge in this work?
For me the biggest challenge was convincing myself and others that we could infer anything about organismal phylogenies from metagenomic data. People had built phylogenies for individual genes from metagenomic data sets, but there was a lot of skepticism about how and whether it would be possible to infer a phylogeny for multiple genes given the short, non-overlapping nature of metagenomic sequences. A post on your blog provided a lot of useful feedback. In the end this challenge was overcome both through the availability of software packages for placement of short sequences onto reference phylogenies, as well as simulation and bootstrap analyses to make sure that the results we were finding were robust.
4. Any additional things left out of the paper that you would like to mention here? Other acknowledgements? Annoyances?
There were a number of people involved in the iSEEM project, including Samantha Risenfeld and Aaron Darling, who did simulations that were very helpful in figuring out when and whether we could make inferences about phylogenetic relationships among metagenomic reads.
Our paper makes use of a large number of open-source software packages and I’d like to thank the people who made their code available for re-use in this way. In particular the short sequence placement methods implemented in packages like RAxML and pplacer made this study possible.
5. What (in general) are your current and future plans?
Right now I’m working at the Biology & the Built Environment Center on a number of projects studying the phylogenetic and functional diversity of microbes in indoor environments, trying to understand the interaction between architectural design and microbial diversity indoors, and the role indoor microbes play in human health and well being. I am still interseted in plant biology, and I have an ongoing project looking at the diversity and function of microbial communities on plant leaves (the ‘phyllosphere’) in tropical and temperate forests.
Kembel, S., Eisen, J., Pollard, K., & Green, J. (2011). The Phylogenetic Diversity of Metagenomes PLoS ONE, 6 (8) DOI: 10.1371/journal.pone.0023214







