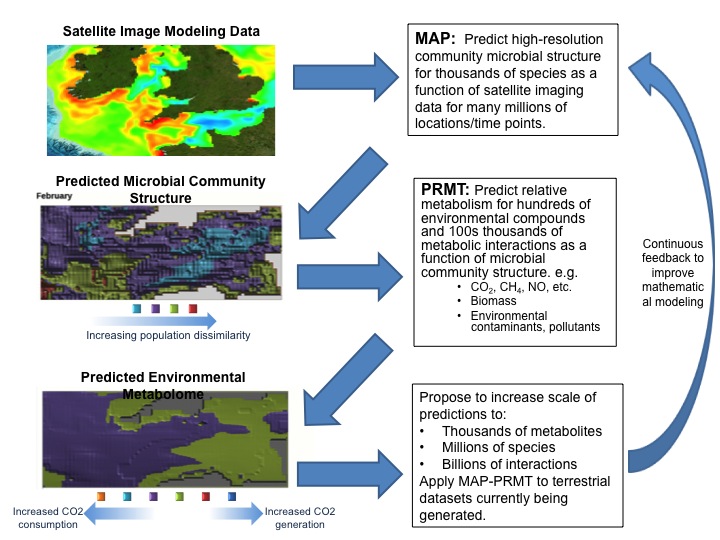
![]()
I note – I found out about this paper from Carl Zimmer who asked me if I had any comments. Boy did I. And Zimmer has a New York Times article today discussing the paper: How Many Species on Earth? It’s Tricky. Here are my thoughts that I wrote down without seeing Carl’s article, which I will look at in a minute.
“Mora et al. [4] offer an interesting new approach to estimating the total number of distinct eukaryotic species alive on earth today. They begin with an excellent survey of the wide variety of previous estimates, which give a range of different numbers in the broad interval 3 to 100 million species”….“Mora et al.’s imaginative new approach begins by looking at the hierarchy of taxonomic categories, from the details of species and genera, through orders and classes, to phyla and kingdoms. They documented the fact that for eukaryotes, the higher taxonomic categories are “much more completely described than lower levels”, which in retrospect is perhaps not surprising. They also show that, within well-known taxonomic groups, the relative numbers of species assigned to phylum, class, order, family, genus, and species follow consistent patterns. If one assumes these predictable patterns also hold for less well-studied groups, the more secure information about phyla and class can be used to estimate the total number of distinct species within a given group.”
| Figure 1. Predicting the global number of species in Animalia from their higher taxonomy. (A–F) The temporal accumulation of taxa (black lines) and the frequency of the multimodel fits to all starting years selected (graded colors). The horizontal dashed lines indicate the consensus asymptotic number of taxa, and the horizontal grey area its consensus standard error. (G) Relationship between the consensus asymptotic number of higher taxa and the numerical hierarchy of each taxonomic rank. Black circles represent the consensus asymptotes, green circles the catalogued number of taxa, and the box at the species level indicates the 95% confidence interval around the predicted number of species (see Materials and Methods). From Mora C, Tittensor DP, Adl S, Simpson AGB, Worm B (2011) How Many Species Are There on Earth and in the Ocean? PLoS Biol 9(8): e1001127. doi:10.1371/journal.pbio.1001127 |
| Figure 2. Validating the higher taxon approach. We compared the number of species estimated from the higher taxon approach implemented here to the known number of species in relatively well-studied taxonomic groups as derived from published sources [37]. We also used estimations from multimodel averaging from species accumulation curves for taxa with near-complete inventories. Vertical lines indicate the range of variation in the number of species from different sources. The dotted line indicates the 1∶1 ratio. Note that published species numbers (y-axis values) are mostly derived from expert approximations for well-known groups; hence there is a possibility that those estimates are subject to biases arising from synonyms. |
| Table 2. Currently catalogued and predicted total number of species on Earth and in the ocean. |
Their approach leads to an estimate of 455 ± 160 Archaea on Earth and 1 in the ocean. Yes, one in the ocean. Amazing. Completely silly too. Bacteria are a little better. An estimate of 9,680 ± 3,470 on Earth and 1,,320 ±436 in the oceans. Still completely silly.
We also applied the approach to prokaryotes; unfortunately, the steady pace of description of taxa at all taxonomic ranks precluded the calculation of asymptotes for higher taxa (Figure S1). Thus, we used raw numbers of higher taxa (rather than asymptotic estimates) for prokaryotes, and as such our estimates represent only lower bounds on the diversity in this group. Our approach predicted a lower bound of ~10,100 species of prokaryotes, of which ~1,320 are marine. It is important to note that for prokaryotes, the species concept tolerates a much higher degree of genetic dissimilarity than in most eukaryotes [26],[27]; additionally, due to horizontal gene transfers among phylogenetic clades, species take longer to isolate in prokaryotes than in eukaryotes, and thus the former species are much older than the latter [26],[27]; as a result the number of described species of prokaryotes is small (only ~10,000 species are currently accepted).
we estimate that there are about 20,000 common species and 500,000 rare species in a small quantity of soil or about a half million species.
We are also able to speculate about diversity at a larger scale, thus the entire bacterial diversity of the sea may be unlikely to exceed 2 × 10^6, while a ton of soil could contain 4 × 10^6 different taxa.
Are their estimates perfect? No surely not. But I think without a doubt the number of bacterial and archaeal species on the planet is in the range of millions upon millions upon millions. 10,000 is clearly not even close. Sure, we do not all agree on what a bacterial or archaeal species is. But with just about ANY definition I have heard, I think we would still count millions.