Jason Stajich speaks Wednesday, June 29th at noon PST on “Fungal phylogenomics: Getting lost in the moldy forest.” Fungi occupy diverse ecological niches in roles from nutrient cycling in rainforest floors to aggressive plant and animal pathogens. Molecular phylogenetics has helped resolve many of branches on the Fungal tree of life and enabling studies of evolution across this diverse kingdom. The genome sequences from hundreds of fungi now permit the study of change in genes and gene content in this phylogenetic context and to connect molecular evolution with adaptation to ecological niches or changes in lifestyles. I will describe our work in studies contrasting pathogenic and non-pathogenic fungi and efforts to unravel the evolution of multicellularity in fungi comparing unicellular basal fungi with multicellular mushrooms and molds. The development of tools for data mining and use of fungal genomics is also driving the pace of molecular biology and genetics of fungi. I will highlight new approaches to make this easier and the ways data integration can inform and transform studies of functional biology of fungi.
Japan
04:00 (04:00 AM) on Thursday, June 30
New Zealand
07:00 (07:00 AM) on Thursday, June 30
West Coast USA
12:00 (12:00 PM) on Wednesday, June 29
East Coast USA
15:00 (03:00 PM) on Wednesday, June 29
England
20:00 (08:00 PM) on Wednesday, June 29
France
21:00 (09:00 PM) on Wednesday, June 29
Learn how to connect ahead of time. To hear about upcoming talks, send an email to phyloseminar+subscribe@googlegroups.com or follow @ematsen. If you can’t make it, don’t fret– you can always watch the recording
I was at a committee meeting yesterday for a great PhD student here at UC Davis, Michael Hornsby and the topic of selfish DNA came up. After his meeting we sat down and looked for some new papers and review papers on the topic. I just thought it might be of value to share some of these here:
———————————— The Call for Lightning Talks is now open for the 2011 conference on Informatics for Phylogenetics, Evolution, and Biodiversity (iEvoBio), athttp://ievobio.org/ocs/index.php/ievobio/2011. See below for instructions.
Lightning talks are short presentations of 5 minutes. They are ideal for drawing the attention of the audience to new developments, tools, and resources, or to subsequent events where more in-depth information can be obtained. Please also see our FAQ for more information ( http://ievobio.org/faq.html#lightning). Lightning talks will be part of the more interactive afternoon program on both conference days.
Submitted talks should be in the area of informatics aimed at advancing research in phylogenetics, evolution, and biodiversity, including new tools, cyberinfrastructure development, large-scale data analysis, and visualization.
Submissions consist of a title and an abstract at most 1 page long. The abstract should provide an overview of the talk’s subject. Reviewers will judge whether a submission is within scope of the conference (see above). If applicable, the abstract must also state the license and give the URL where the source code is available so reviewers can verify that the open-source requirement(*) is met.
Review and acceptance of lightning talks will be on a rolling basis. The deadline for submission is the morning of the first day of the conference (June 21). Note that the number of lightning talk slots is finite, and given the high volume of submissions we experienced for full talks, the Lightning Talks track may fill up early. We cannot accept lightning talks until the open-source requirements are met, and so waiting with that until the deadline risks that the track is full by that time.
We ask all submitters of lightning talks to be willing to also serve as reviewers of such, as described above.
Lightning talks are only 1 of 5 kinds of contributed content that iEvoBio will feature. The other 4 are: 1) Full talks (closed), 2) Challenge entries, 3) Software bazaar demonstrations, and 4) Birds-of- a-Feather gatherings. The Call for Challenge entries remains open (see http://ievobio.org/challenge.html), and information on the Software Bazaar and Birds-of-a-Feather sessions is forthcoming.
More details about the program and guidelines for contributing content are available at http://ievobio.org. You can also find continuous updates on the conference’s Twitter feed athttp://twitter.com/iEvoBio.
iEvoBio is sponsored by the US National Evolutionary Synthesis Center (NESCent) in partnership with the Society of Systematic Biologists (SSB). Additional support has been provided by the Encyclopedia of Life (EOL).
The iEvoBio 2011 Organizing Committee: Rob Guralnick (University of Colorado at Boulder) (co-Chair) Cynthia Parr (Encyclopedia of Life) (co-Chair) Dawn Field (UK National Environmental Research Center) Mark Holder (University of Kansas) Hilmar Lapp (NESCent) Rod Page (University of Glasgow)
(*) iEvoBio and its sponsors are dedicated to promoting the practice and philosophy of Open Source software development (see http://www.opensource.org/docs/definition.php) and reuse within the research community. For this reason, if a submitted talk concerns a specific software system for use by the research community, that software must be licensed with a recognized Open Source License (see http://www.opensource.org/licenses/), and be available for download, including source code, by a tar/zip file accessed through ftp/http or through a widely used version control system like cvs, Subversion, git, Bazaar, or Mercurial.
A semi quick one here. I am writing this in part because it is really a lot of fun to be at UC Davis with all the excellent evolution and ecology stuff going on here. Some links for those who might be interested in learning more about Evolutionary studies at UC Davis include:
There is more but that is a good start. Anyway a recent press release from Davis caught my eye in part because I know the people involved and also in part because I was unaware of the details of what they have been working on. The press release is titled “Explosive Evolution in Pupfish” and discusses some interesting research by a PhD student Chris Martin and his advisor, my colleague Peter Wainwright. The work was published in Evolution and is entitled: “TROPHIC NOVELTY IS LINKED TO EXCEPTIONAL RATES OF MORPHOLOGICAL DIVERSIFICATION IN TWO ADAPTIVE RADIATIONS OF CYPRINODON PUPFISH” (DOI: 10.1111/j.1558-5646.2011.01294.x). Alas it is not OpenAccess, but the paper is available on their lab web site here.
The work is a bit out of my arena, and I suppose I could critique the press release a bit, but I won’t right now. As a side note, I should mention I really love pupfish so that also caught my eye, and I have occasionally tried to convince Chris to look at the microbes in pupfish.
Anyway, rather than bore people with my thoughts, I thought it might be nice to post some comments I got from Chris about the paper. I got these is a series of emails and though they are a bit out of context, I am just going to post them here:
Note that the press release is a bit confusing: there are other scale-eating fishes (has evolved at least 14 times independently), but this is the only scale-eating pupfish (and only scale-eater among all 1500 atherinomorphs).
#2: Pupfish are indeed named after puppy dogs for their playful swimming behavior!
#3: I think the most exciting thing about this system is that it presents the opportunity to study the origins of ecological novelty in a very recent radiation (possibly as young as 8,000 years if we go by geographic dates of the lakes). This study leaves many outstanding questions that I hope to address in my future research.
For example, why does exceptional adaptive radiation occur on these two islands and nowhere else in the Caribbean? Is this due to lack of sampling, is there something unique about these two environments, or is there something unique about the founding populations in these two cases? Both lakes are large, isolated, productive environments with only 1 or 2 other competing fish species and this is surely part of the story. But, there are many other large lakes in the Caribbean, often with very similar fish communities. Further, note that the other competing fish species have not diversified at all: is this due to their time of arrival or is there something special about pupfishes? I’m currently planning to do broader sampling of pupfish populations and lake environments across the Caribbean to address these questions.
Second, what factors actually drive such dramatic rates of morphological diversification? I have just returned from a trip to San Salvador Island where I setup four field enclosures and added juvenile pupfish to estimate a fitness landscape for jaw morphology in this environment. Juveniles were F2 hybrids of the three species raised in the lab here at Davis in order to sample from the full spectrum of phenotypic variation. I will be returning in July to collect this experiment and I do hope my enclosures and some fish survive! This study should provide an estimate of the strength of selection on existing phenotypes as well as potentially unfit intermediate phenotypes.
Finally, why have different sets of resource specialists evolved in very similar environments? In particular, why has a specialized scale-eater failed to evolve in Mexico – there are obviously scales to feed on and the fish densities appear comparable. Scale-eating has evolved independently many times, but why don’t all fish communities contain scale-eating specialists?
Anyway, going to try to write more about Evolutionary studies at UC Davis in the future. I am always amazed at how much interesting work there is here.
I note this is a general post about the trip – I will post more about the individual science topics including C-DEBI and Virgin Oceanic later.
As is usual, I did not fully commit to going to the workshop immediately and I dragged out committing for a very long time (driving Bill I am crazy I am sure). But eventually I accepted and then kept flip-flopping on exactly when I would go, but eventually settled on dates too.
What is C-DEBI:
When Bill first invited me to this workshop, I had no clue what this C-DEBI project was. And Bill must have assumed I knew because he did not provide any detail about what C-DEBI was. So of course, that is what that Google thing is for. And what I found was quite intriguing:
Welcome to the Center for Dark Energy Biosphere Investigations (C-DEBI), a National Science Foundation (NSF)-funded Science and Technology Center on the deep biosphere. Our mission is to explore life beneath the seafloor and make transformative discoveries that advance science, benefit society, and inspire people of all ages and origins. We are a multi-institutional distributed center establishing the intellectual, educational, technological, cyber-infrastructural and collaborative framework needed for transformative experimental and exploratory research on the subseafloor biosphere.
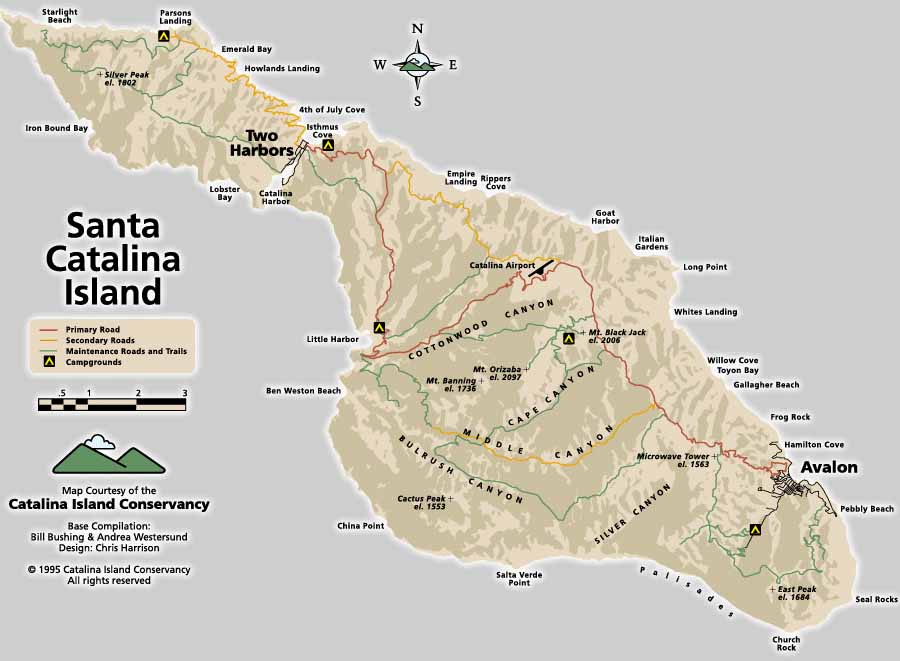
This certainly intrigued me. And the fact that the workshop was going to be at the George and MaryLou Boone Center for Science and Environmental Leadership (which is part of the USC Marine Station on Catalina Island) also appealed – I had visited the Marine Station on Catalina Island in the summer of 2009 for a week and it was very very very nice.
So I kind of had to say yes. Rough I know – being forced to go to a meeting on Catalina Island because my kids had gotten a great submersible tour.
Heading to Catalina Island
So I finally got my act together and headed down to LAX from Sacramento.
I arrived in LAX and got a cab to the Catalina Island Ferry terminal. I picked up my ticket and alas, the deli there was closed and there was nowhere to get lunch. I wandered around a bit and took some pictures.
As a bit of a side story, a PhD student in my lab Russell Neches was visiting his mom in the LA area and he and his mom dropped by for a few minutes. Then Katrina and her daughter were dropped off by her husband Eric Webb (who does some interesting marine microbiology research himself).
After saying goodbyes, we boarded the ferry and headed out. Katrina and I were both pleased (and surprised) to hear the announcement that we were going to Two Harbors first, rather than Avalon, so we would get there much earlier.
We headed out into San Pedro harbor, slowly, and I and everyone else took pics as we went by some of the sights.
It is always amazing to me to see the giant container ships and the massive size of San Pedro Harbor.
The pirate ship was a bit weird, but I guess it must be some sort of tour thing.
Then we got out into the more “open” water. The seas were pretty small – but the ferry goes quite fast so it was bouncing up and down a little bit. Unlike on my last trip, when we say a few blue whales from the ferry, we did not see much animal life in the water.
We got the Catalina Island pretty quickly. And it was looking gorgeous – much greener than the last time I was there. And we passed by the Marine Station – and headed to the dock (see the panoramic pictures I made using Adobe Photoshop’s stitching function):
Some of the C-DEBI personnel picked us up in town and we headed up/down the dirt road in the USC Van to the marine station. We got there and I found out I was in the same townhouse/apartment I had stayed in in 2009. Nice. After dumping my stuff there was a reception in the Boone Center. I got to meet the rest of the people there for the meeting – it was a small collection of folks. We had some wine and cheese and other goodies, enjoyed the view of the lab and the water and then headed over to the dining hall for dinner.
After dinner we went back to the Boone Center and spent the night telling stories and getting to know each other and the Woolly Bear caterpillars wandering around everywhere.
Katrina Edwards and wooly bear
Katrina Edwards
I went to sleep and got up semi-early the next AM. I made myself some coffee and headed to breakfast. Then down to the lab for a full day of meetings and discussion. But first I took a look around and took a few pictures:
This was when I finally got a better introduction to the whole point of the meeting. The point of the meeting was that Bill Nelson was tasked with organizing a “theme” for the C-DEBI project on evolution. In essence, our meeting was to discuss what interesting evolution-related questions could be asked/answered as part of the C-DEBI project.
Basically, just as the meeting started we all headed down to the dock to welcome the arrival of some other folks from the mainland. The new arrivals were Ann Close and two members of the Virgin Oceanic Project – Chris Welsh and Loretta Whitesides. This project was announced very recently and is a project to explore the five deepest sites in each major ocean in a one person submersible. The pilot of the submersible will be Chris Welsh. The project is being supported in part by Sir Richard Branson and thus the “Virgin” connection. The sub is being designed by Graham Hawkes a well known ROV designer. More on this in a bit. I note I had written about this in my blog a few days ago –— not knowing I would soon be meeting some of the people involved.
We then headed back to the meeting room (the library) and got going. We did mini introductions. At the suggestion of Chris Welsh, everyone in addition to saying who they were also said who their hero was. Among the people listed were relatives of participants, Charles Lindberg, Yoda, Charles Darwin, superheroes, and oceanographers. We then got a more detailed introduction to the C-DEBI project and also got a very brief introduction to the Virgin Oceanic Deep-Five project (more on this below).
We then had a mini coffee break and I somehow handed over digital SLR my camera to Katrina’s daughter. She then took it and generated quite a collection of good pictures of the people at the meeting.
Jonathan Eisen
Bill Nelson
Katrina Edwards
Loretta Whitesides
Jennifer Biddle
Jason Sylvan
Craig Moyer
Billy Brazelton
Chris Welsh
Ben Tully
John Heidelberg
We then had some additional discussions about evolution and the C-DEBI project. I learned, for example, about a group of bacteria I alas had not heard of before – the Zeta proteobacteria (see Moyer’s PLoS One paper on them here). This is a group that is particularly abundant in some C-DEBI related sites. In particular they seem to do well in iron-oxidizing microbial mat communities. Moyer presented some interesting data on biogeography of this group of bacteria. I also learned some new things from the others at the meeting. And then we broke for lunch up the hill in the dining hall.
After lunch we hear much more detail on the Virgin Oceanic project. It is completely fascinating, though a bit scary. The plan is for Chris Welsh to pilot the submersible down into the deepest sites on each of the five main oceans. Right now the submersible is still being finished. We also learned about the sailboat that will be the mother ship for the submersible. The boat seems quite fast and has some nice features but it will also be a bit tight on space. I note Welsh mentioned they are still looking for crew for the boat so if you are interested …
Their plan is to do some testing in a few months in various places and to then do the deep dives. What was most interesting to me about the project is that the people involved really seem to be committed to doing interesting science. Loretta Whitesides has a science background and seemed to have an excellent grasp of many of the scientific issues being discussed. Welsh also seems to have a deep interest in the science. The group also has some good people lined up that they are working with and are still looking for other ideas and collaborators to participate in the science. It reminded me of some of the stories I have heard about the great explorer’s doing science along the course of their voyages.
Then back for some more meeting and discussion. During the course of the day I learned about an enormous number of cruises and surveys and plans for drilling in various sites and how the C-DEBI folks study microbes beneath the sea floor. I also learned that they have a lot of education and outreach activities and are looking for more. I also learned that if you want to keep up to date on C-DEBI related activities and if you want to participate in some of their projects, they are very open. A good way to keep up to date is to join their mailing list. One can also learn a great deal by browsing their web site and some of the publications listed there. Anyway – I am going to do a whole post just on C-DEBI later — focusing here on the big picture parts of my trip.
After the discussions we went back to Boone House for another reception and then dinner. After dinner we hung out in the Boone House again. And then went to sleep (though there were rumors of some weird sightings in and around the housing complex that night.
The next morning I got up a bit late and missed breakfast but I grabbed some cereal from the dining hall, made some coffee and headed down the hill again. We had some discussions in the AM about the future plans for evolutionary studies associated with the C-DEBI project and then headed back up the hill for lunch. There was a little bit of time before lunch so I wandered around the hills and took a few pictures.
Then we had our last group lunch and many of the folks headed down to the USC boat to get back to the mainland.
I spend the next few hours doing a bit of work and also went to the beach to collect some shells for my kids. You see, I did not have to get on the USC boat because I had snagged a ride on a helicopter back to the mainland.
The helicopter eventually arrived and Katrina gave me a ride down to the water in her golf cart (she had recently injured her leg and had a hard time walking around).
And we got in the chopper and were off. Katrina’s daughter and John and Karla Heidelberg’s son enjoyed the ride quite a bit. It was my first helicopter ride too – and it was quite fun. The best part the pilot saw and then flew over a massive pod of dolphins.
And then we headed on into San Pedro (which freaked me out a bit as they had said we were going to Long Beach and I had to catch a plane).
John Heidelberg however had figured this out and met us there. The chopper took off, Katrina and her daughter went to wait for her husband, and John gave me a ride to the airport and I got there in time for my flight home.
This one is definitely worth a read for phylogeneticists and phylogenomicists (is that a word?) out there: PLoS Biology: Resolving Difficult Phylogenetic Questions: Why More Sequences Are Not Enough. Philippe et al. discuss some important issues in using genomes to infer phylogenies of species in this commentary/review paper. They discuss in particular some recent studies of animal evolution but they cover a lot of useful ground here and include a good review of terminology and some of the basic issues at play. I am personally going to have to read it in more detail to help deal with some of the issues in our recent study of novel sequences in metagenomic data.
This is a post about a paper that has been a long long time coming. Today, a paper of mine is being published in PLoS One. The paper is titled “Stalking the Fourth Domain in Metagenomic Data: Searching for, Discovering, and Interpreting Novel, Deep Branches in Marker Gene Phylogenetic Trees” and is available at http://dx.plos.org/10.1371/journal.pone.0018011. (or if that link does not work you can get a copy here). This paper represents something I started a long time ago and I am going to try to describe the story behind the paper here.
I note – we are not doing a press release for the paper, for a few reasons. But one of them is that, well, I am starting to hate press releases. So I guess this is kind of my press release. But this will be a bit longer than most press releases. I note – my key fear here is that somehow in my communications with the press or in our text in the paper or in this post I will overstate our findings. Here is the punchline – we found some very phylogenetically novel forms of phylogenetic marker genes in metagenomic data. We do not have a conclusive explanation for the origin of these sequences. They may be from novel viruses. The They may be ancient paralogs of the marker genes. Or they may be from a new branch of cellular organisms in the tree of life, distinct from bacteria, archaea or eukaryotes. I think most likely they are from novel viruses. But we just don’t know.
UPDATE: Am posting some links here to news stories/blogs about our paper
In the paper, we searched through metagenomic data (sequences from environmental samples) for phylogenetically novel sequences for three standard phylogenetic marker genes (ss-rRNA, recA, rpoB). We focused on sequences from the Venter Global Ocean Sampling data set because, well, we started this analysis many years ago when that was the best data set available (more on this below). What we were looking for were evolutionary lineages of these genes that were separate from the branches that corresponded to the three known “Domains” of life (bacteria, archaea and eukaryotes).
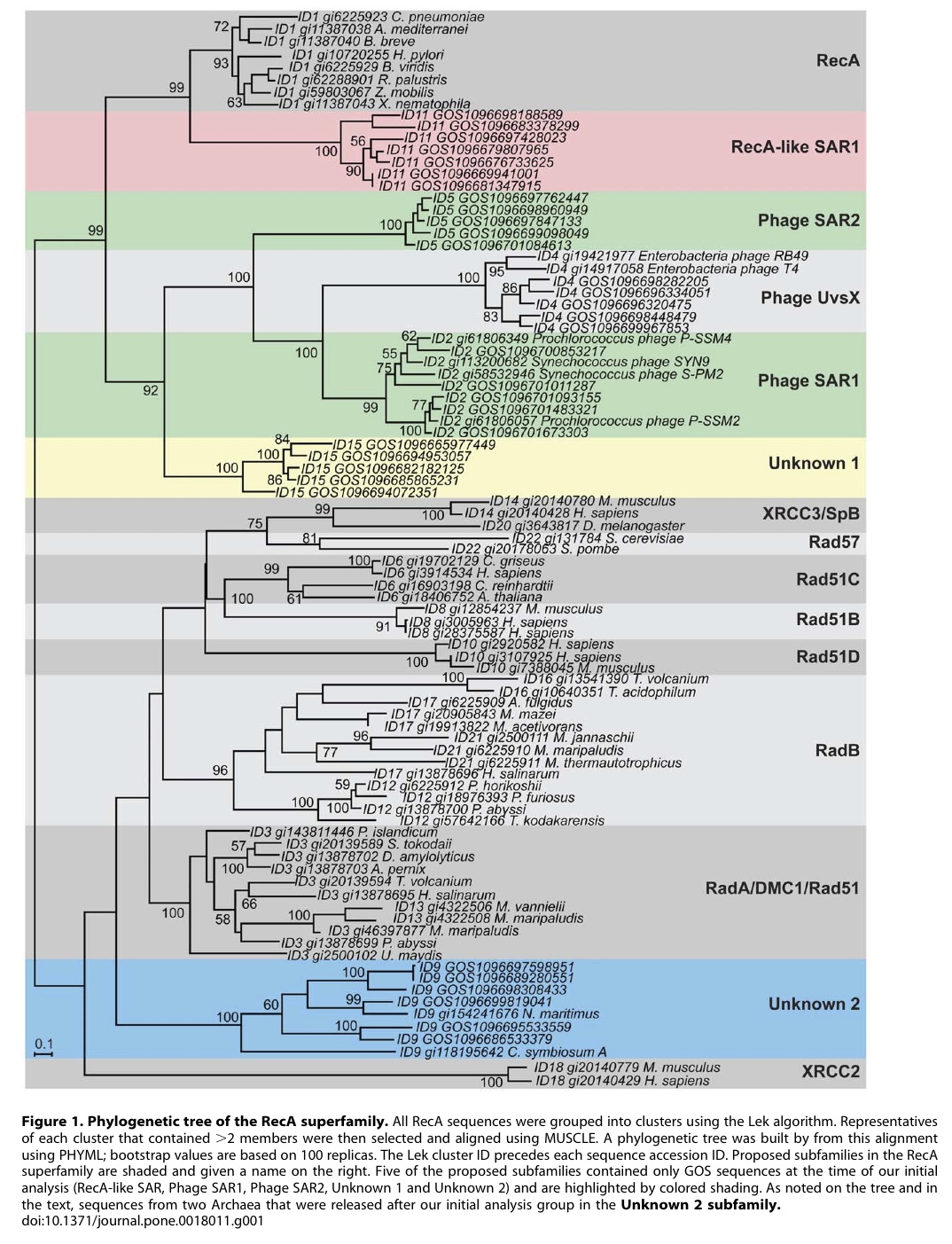
To search for such novel lineages in the metagenomic data, we built evolutionary trees using these genes where we included sequences from known organisms (and viruses) as well as sequences from metagenomic data. We then looked through the trees for groups that were both phylogenetically novel and included only environmental data (i.e., they were new compared to known organisms or viruses). This method did not work very well for rRNA sequences (largely because making high quality alignments of short phylogenetically novel rRNA sequences was difficult – more on this below). But with RecA and RpoB homologs we were able to generate what we believe to be robust phylogenetic trees. And in these trees we found evidence for phylogenetically very novel sequences in environmental data.
Figure 1. Phylogenetic tree of the RecA superfamily.
Figure 3. Phylogenetic tree of the RpoB superfamily
We then propose and discuss four potential mechanisms that could lead to the existence of such evolutionarily novel sequences. The two we consider most likely are the following
The sequences could be from novel viruses
The sequences could be from a fourth major branch on the tree of life
Unfortunately, we do not actually know what is the source of these sequences. So we cannot determine which of the theories is correct. Obviously if there is a novel lineages of cellular organisms out there, well, that would be cool. But we have no evidence right now if that is what is going on. Personally, I think it is most likely that these novel sequences are from weird viruses. But as far as we can tell, they truly could be from a fourth major branch of cellular organisms and thus even though we did not have the story completely pinned down, we decided to finally write up the paper to get other people to think about this issue.
Below I give all sorts of other details about the project in the following areas
The history of the project
More detail on what is in the paper
Follow up analysis and rapid posting with google Know
Data deposition in Dryad
Who was involved
UPDATE: Funding for this work
The history of the project
Well, this is one of those projects for which the history is hard to explain. We started this work in 2004 when I was helping Venter and colleagues analyze the Sargasso Sea metagenome data. I was working at TIGR in 2003, which are the time was a sister institute to some of the institutes affiliated with the J. Craig Venter Institute (JCVI) (it was a complicated time). Craig had led a project to do a massive amount of shotgun sequencing of DNA isolated from the Sargasso Sea, which had been the site of many previous studies of uncultured microbes. And Craig, as well as some of the people working with him including John Heidelberg who was at TIGR, had asked me to help in analysis of the data. So I eventually went to a meeting about the project and got involved. It was quite exciting and I put a lot of effort into helping analyze the data.
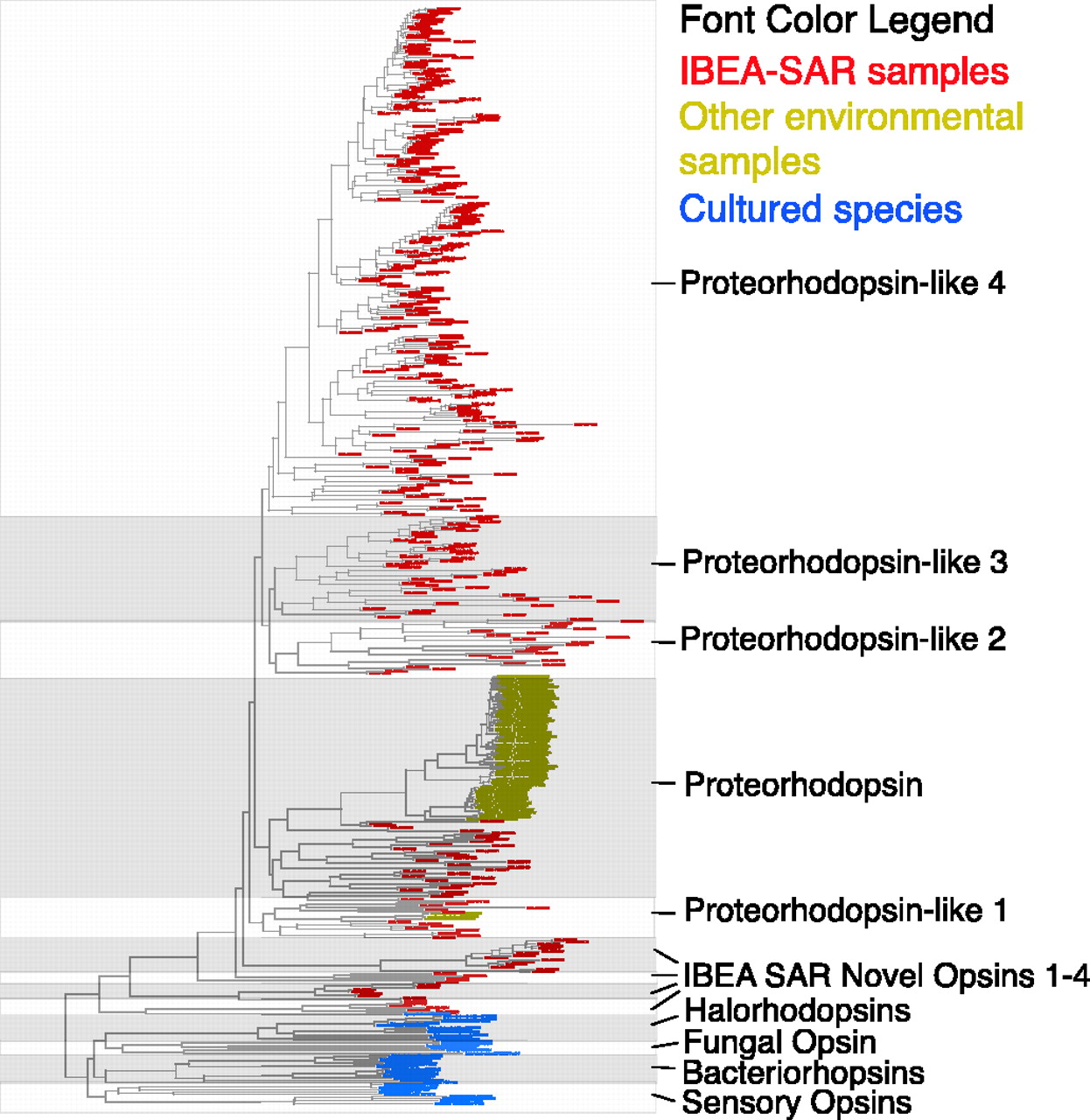
As part of my work on the project, I and Martin Wu and Dongying Wu did a variety of phylogenetic studies of genes and gene families. One of these, was a phylogenetic analysis of proteorhodopsin homologs showing massively more diversity in the Sargasso data than in the PCR experiments done by Delong and Beja and others.
Figure 7 from Venter et al. 2004.
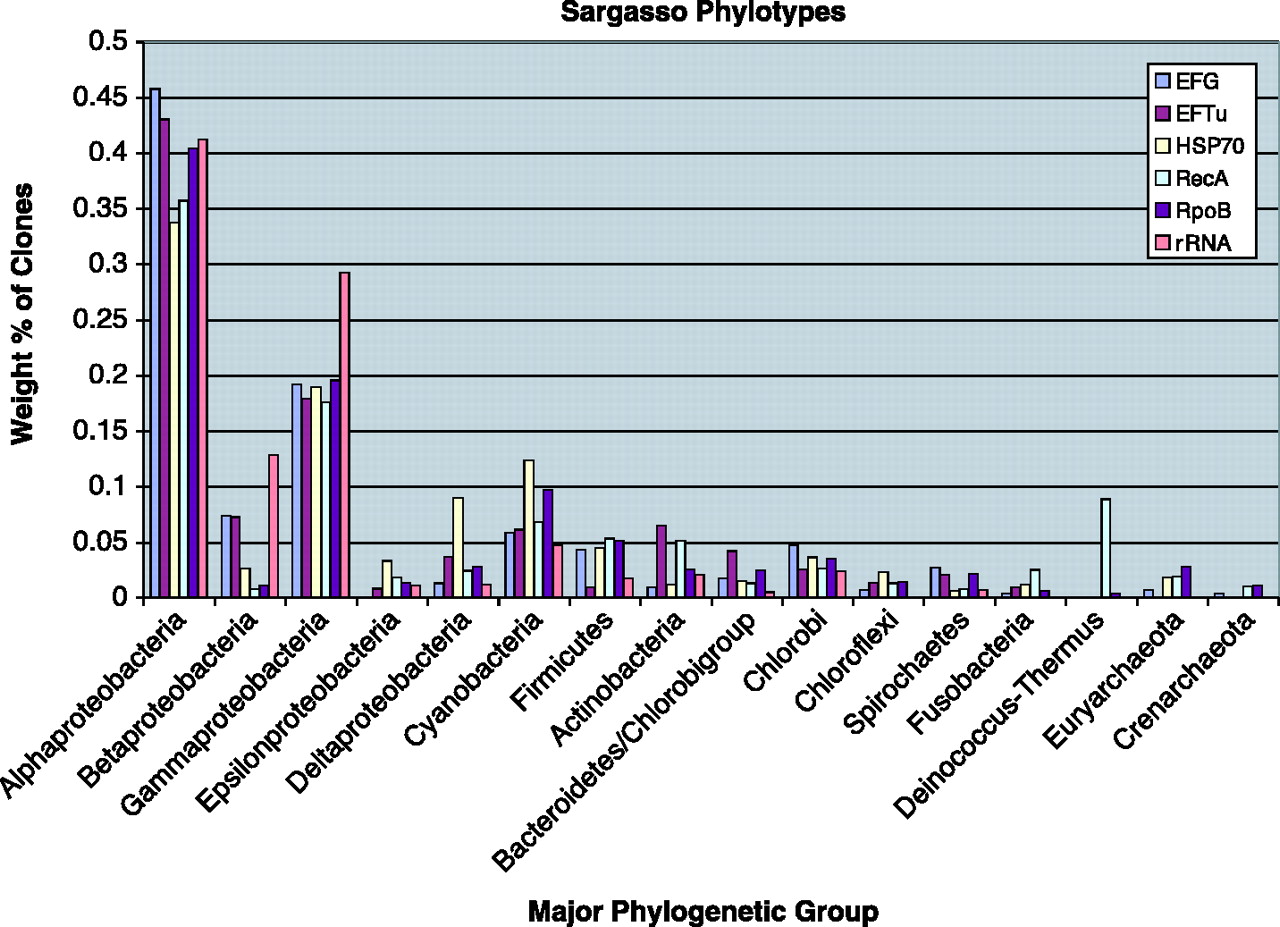
We also did the first “phylotyping” in metagenomic data using genes other than rRNA. We built trees of bacterial ss-rRNAs, RecAs, RpoBs, HSP70s, EF-Tus and EF-Gs and then assigned each sequence to a phylum from the trees. In this analysis we found a variety of interesting things.
Figure 6 from Venter et al. 2004.
One thing I did not include in the Sargasso paper was an analysis I did of RecA homologs where I tried to include ALL RecA-like genes from bacteria, archaea, eukaryotes and viruses. The trees I made were a bit unusual but I was not sure that the alignments I had made were robust or that I had found all the RecA-like genes of interest so I did not even show this to Craig et al. at the time.
UPDATE: I note – our work on this project was supported by a grant from the NSF Assembling the Tree of Life program that was awarded to me and Naomi Ward and Karen Nelson. Those funds supported the development of many of the informatics tools we used in this analysis and Martin and Dongying were both working on that project.
After the Sargasso paper was published in 2004 though, I continued to fester about the RecA trees. And I wondered – if instead of trying to classify bacterial sequences into phyla, what if I tried to look for RecAs, rRNAs and other genes that were completely new branches in the tree of life? I got the chance to start to play with this concept again when Venter and crew asked me to help analyze the data coming out of the Global Ocean Sampling project. Again, this project was very exciting and interesting.
As part of the project, I helped Shibu Yooseph and others look into whether the GOS data revealed any completely new types of functionally interesting genes, much like I had shown for proteorhodopsin in the Sargasso data.
Figure 7 from Yooseph et al. 2007 . Phylogenies Illustrating the Diversity Added by GOS Data to Known Families That We Examined
And again my mind started wandering towards the question of “OK – so – if there are all these very unusual and novel functionally interesting genes, what about looking for unusual and very novel phylogenetic marker genes”? So finally, I got back to work on the issue.
And so I built a better RecA tree by first pulling out all possible homologs of RecA and RecA like proteins from the GOS data and then building an alignment and a tree. And there they were. Some very f*%&$ novel RecAs – distinct from any previously known RecA like proteins as far as I could tell. And so with help from Dongying and the JCVI crew, we started building a story about novel RecAs. And then we looked at RpoBs. And found novel ones too. And in mid 2006 while Shibu and Doug worked on their papers that were to be submitted to PLoS Biology and I worked on a review paper too, I told Emma Hill (who has since changed her name to Emma Ganley due to some sort of wedding thing) at PLoS Biology about the an analysis that was consistent with the existence of a fourth domain of life. No overstating our findings really – just that we found very novel phylogenetic marker genes. And that I was working on a paper on it. But alas I never got it done, though I was happy to have convinced Venter to send the GOS papers to PLoS Biology and I think the papers that came out were good. Among the papers were my review (Environmental Shotgun Sequencing: Its Potential and Challenges for Studying the Hidden World of Microbes, Doug Rusch’s diversity paper The Sorcerer II Global Ocean Sampling Expedition: Northwest Atlantic through Eastern Tropical Pacific and Shibu’s protein family paper The Sorcerer II Global Ocean Sampling Expedition: Expanding the Universe of Protein Families as well as many others as part of the Ocean Metagenomics Collection at PLoS.
And in the midst of all of this, we had our first child and we wanted to move back to Northern California to be closer to family (my wife’s family is all in the Bay Area and my sister and brother Michael were in N. Cal too). So I applied for jobs and eventually took at job at UC Davis and we moved to Davis. Needless to say, all of that put a bit of a crimp in my work productivity. And once I was up and running at Davis, it just took a long time to get back to the searching for novel deep branches in the tree of life. But finally, we did it (with periodic prodding from Craig Venter). And we put together a paper and got it submitted to PLoS One in October. The reviews were very positive and enormously helpful. And we finally got a revision in January and it was officially accepted in February 2011. Only some seven years after my first work on the project. Whew.
More detail of what is in the paper Well, I am going to be posting here some additional detail on what is in the paper.
Why we punted on analysis of very novel rRNAs.
The problem with rRNA is that the sequences that come from environmental samples are not complete (i.e. they only correspond to portions of the rRNA genes). Unfortunately, this makes a key step in phylogenetic analysis difficult – the alignment of sequences. We actually found about 200 rRNA sequences that seemed unusual in a phylogenetic sense. However, we were not convinced that the alignments of these fragments to other rRNAs was robust. This is because the alignment of rRNAs is best done making use of the base pairing secondary structure of the molecule and not the base sequence (i.e., primary structure).
With only rRNA fragments, we could not use the secondary structure to do the alignments because you need to whole molecule to determine the best folding. Combined with the fact that we were searching for very distantly related ribosomal RNAs which would be hard to align even if we had the whole molecule, we were stuck for a bit. It seemed impossible to look for really novel organisms. So that is when we turned to other genes. The key for this is that there are protein coding genes that are universal and that for known organisms show similar patterns to rRNA in trees. In fact, in 1995 I wrote a paper showing that trees of RecA were very similar to trees of rRNA. RpoB is also considered a very robust phylogenetic marker. For organisms that we have in the lab (i.e., cultured) – many people use these other genes for phylogenetic analysis. rRNA has been very important in part because of the ease with which one can PCR amplify it from environmental samples and the fact that it is very hard to PCR amplify protein coding genes from the environment. Metagenomics changes this. With random sequencing, you get data from all genes. This means we can pick and choose genes to analyze for phylogenetic analysis and do not have to rely on rRNA.
So we went after RecA first, because it has been shown to be a good phylogenetic marker for studies of the tree of life. And we found some very novel branches in the RecA tree. And after analyzing these and convincing ourselves that they were indeed phylogenetically very novel we went after RpoB. And also found very novel branches.
So the phylogenetic analysis I think is very robust.
RecA and RpoB as phylogenetic markers
Many genes have been used as alternatives to rRNA genes to build “Trees of Life” including all organisms. Each has their own flavors of advantages and drawbacks. Two commonly used ones are the RecA and RpoB superfamilies.
The many possible explanations for finding novel forms of phylogenetic marker genes
The phylogenetically novel phylogenetic marker genes we found could have many explanations including that they could be ancient paralogs of these genes (but not found in any genomes we have available), they could be from viruses, or they could be from a novel branch on the tree of life. Or our trees could be bad. We think the latter is somewhat unlikely as our analysis has many lines of support. For example our RecA trees are very similar to those from a comprehensive study from M. Nei’s lab except they did not include the metagenomic data. But I guess it is still a possibility that our trees are biased in some way (e.g., by long branch attraction or bad alignments)
Follow up analysis and rapid posting via Google Knol
Amazingly and a bit sadly, I think we rushed the paper out. We left out one thing partly by accident – we had done an analysis of the locations from which these novel RecA and RpoB sequences had come. And somehow, in our final push to get the paper out, we left this out. I will be posting this information as soon as possible here and on the PLoS One site.
In addition, after submitting the revision of our paper, we realized that we might be able to do a deeper analysis on one aspect of the work – how RpoB homologs from unusual DNA viruses compared to our novel sequences. We had included some RpoBs from DNA viruses in our analyses but not all that were available. So Dongying Wu did a very rapid additional analysis, adding some additional RpoB homologs to our alignment and making a tree of them. We then wrote a Google Knol about this new tree and submitted the Knol to PLoS Currents “Tree of Life” where it is currently in review. We are publishing the preprint of this Knol to make it available to all even while it is in review.
Figure 2 from Wu and Eisen submitted.
Data availability
There is a move afoot to make sure all data/tools associated with publications are readily available. We used publicly available sequence data and as much as possible publicly available tools for our work . We are trying to release as much as possible to allow people to re-analyze our work and to do any of the work themselves. We have therefore made use of the Dryad Data deposition service to post some of this material (see http://datadryad.org/handle/10255/dryad.8385).
Who was involved
Dongying Wu a brilliant “Project Scientist” in my lab led the project (Project Scientist is one of the UC positions that is like what others call “Senior Scientist”). Dongying is simply one of the best bioinformaticians/computational biologists I have ever met. He was first author on many key papers from my lab including the Genomic Encyclopedia paper that came out last year and the glassy winged sharpshooter symbionts paper that came out a few years ago. Dongying worked in my group at TIGR and moved with me to UC Davis and currently splits his time between UC Davis and the DOE Joint Genome Institute.
Martin Wu. Martin is an Assistant Professor at the University of Virginia. Prior to that he was a Project Scientist in my lab at Davis and a post-doc in my lab at TIGR. He is also a phenomenal bioinformatician / computational biologist. He developed the AMPHORA software in my lab and also led many genome projects (back when sequencing a genome was hard …) including that of the first Wolbachia genome and that of a very unusual bug Carboxydothermus hydrogenoformans. Martin helped with some of the genome analyses as part of this work.
Aaron Halpern, Doug Rusch and Shibu Yooseph are all bioinformaticians from the J. Craig Venter Institute (Aaron is no longer there). All three helped with different aspects of dealing with and analyzing the GOS data and all three have been remarkably patient as this work dragged on and on.
Marv Frazier from the JCVI was helpful in the initial set up and conceptualization of the project.
J. Craig Venter is, well, Craig Venter, and he was involved in multiple aspects of the project including thinking about how and where to look for unusual sequences and interpreting some of the results.
UPDATE: Funding for this work
Most of my labs early work on this project was supported by a grant we had from the Assembling the Tree of Life program at the National Science Foundation (grant 0228651 to me and Naomi Ward). In that project we were working on sequencing and analyzing genomes from phyla of bacteria for which genomes were not available at the time. As part of this work we were designing methods to build phylogenetic trees from metagenomic data because we thought that our new genomes would be very useful in helping analyze metagenomic reads and figure out from which phyla they came. Later work on the project was supported by a grant to me, Jessica Green and Katie Pollard from the Gordon and Betty Moore Foundation (grant 1660).
Some questions that might be asked and some answers (based in part on questions I have gotten from reporters). Note if you have other questions please post them here or on the PLOS One site for the paper.
Why no press release? Well, in part, because I sent information too late (shocking I know) to the Davis Press Office. But also because they have gotten suddenly busy with some Japan earthquake related actions. But also because, well, I really hate a lot of press releases. And finally, my brother had dinner with Carl Zimmer recently and apparently they discussed the possibility of having no press releases associated with papers. So here goes ….
Really – what took so long? I would like to say the US Government made us hold back on publishing this until they could look into whether Venter collected ocean data from Roswell, NM or not. But really, the story above is true. We just did not get it done earlier.
Why do you not know the source of the DNA (i.e., cells, viruses, etc)? This is why there was a six year wait between discovery and writing this up. We kept thinking we would be able to find the organisms but since I moved from TIGR and started a new job, we just never got around to getting to the source. We therefore decided to open this up to others who will hunt for the source by writing up the paper.
Why did you not rename the Unknown 2 group in the RecA tree? We should have renamed our group “Thaumarchaeota” or something like that. When we did the initial analysis our group was novel. And then a few years ago a few groups obtained data from what is thought to be the third major lineage of Archaea – referred to by some as Thaumarchaeota. This is to go with the Euryarchaeota and Crenarchaeota. See http://www.ncbi.nlm.nih.gov/pubmed/20598889 for example.
One of the clades in the RecA tree (XRCC2) seems out of place phylogenetically. I can see how that is confusing. The XRCC2 clade is very weird and hard to figure out. It is not the “normal” eukaryotic genes – those are the Rad51/DMC1 genes. One complication with the RecA family is that there have been duplication events to go with the species evolution. And thus eukaryotes have Rad51, DMC1, Rad51B, Rad51C, Rad57, XRCC3 and XRCC2. We tried to figure out where the XRCC2 group should go but it just was hard to place. The statistical support for its position (we used a method called bootstrapping) is low (note the lack of a number on the node where the branch leading to XRCC2 connects to the base of the tree). Most likely that group should be placed with some of the other eukaryotic groups. However, it seems likely that there was a duplication in the lineage leading up to the ancestor of eukaryotes and archaea (some studies have indicated they share a common ancestor to the exclusion of bacteria). Such a duplication would explain why basically all archaea have a RadA and and RadB and all / most eukaryotes have multiple paralogs as well.
The Unknown 1 group in the RpoB RecA tree seems to group with phage. What can you say about that? We think unknown 1 is potentially of viral origin but still cannot tell. The fact that is clusters with RecA superfamily members from phage suggests this but it is distant enough from known phage for us to not be confident in any predicted origin. As for derivative forms vs. independent branch – this is one of the big questions about viruses these days. Many viruses encode homologs of “housekeeping” genes found across bacteria, archaea and eukaryotes. And in many cases the viral versions of these genes appear to phylogenetically very novel. This is why the people studying mimivirus (which we refer to) suggest some viruses may in fact represent a fourth branch on the tree of life. It is possible that some viruses are in fact reduced forms of what were once cellular organisms – akin to parasitic intracellular species of bacteria possibly.
Why are these phylogenetically novel sequences so low in abundance? This is a key question. I think it would be easy to come up with a theory for these being rare or these being common. They might be rare if their niche is very limited today. Or they might be rare because they could not be very competitive with other organisms. Or they could be rare because they require some unusual interactions with other taxa. In addition, we have only looked carefully at ocean water samples. If these are common somewhere else (e.g., hotsprings, deep subsurface, etc) we would not yet have figured that out. We are looking at additional metagenomic data right now to see fi we can find any locations where relatives of these genes are more common
Dongying Wu, Martin Wu, Aaron Halpern, Douglas B. Rusch, Shibu Yooseph, Marvin Frazier,, & J. Craig Venter, Jonathan A. Eisen (2011). Stalking the Fourth Domain in Metagenomic Data: Searching for, Discovering, and Interpreting Novel, Deep Branches in Marker Gene Phylogenetic Trees PLoS One, 6 (3) : 10.1371/journal.pone.0018011
Theoretical and Empirical Issues in Putting Dates on Phylogenies”
Featuring a Diverse and Distinguished Panel of Discussants
Followed by vigorous audience discussion
Panel members representing different approaches will give short informal presentations (10 minutes each), to be followed by active audience participation (this all following traditional pizza and beer, of course!).
Confirmed Panel Members:
Tracy Heath
Pat Holroyd
Nick Matzke (moderator)
Sarah Werning
The venerable Biosystematists group (http://www.biosystematists.org/), operating since 1936 (see the history on the website), is the only inter-institutional seminar/discussion group on evolution for the Bay Area, so we encourage everyone to join in.
Schedule and venue:
5:30 – social gathering with beverages (beer and soft drinks) and informal pizza dinner: cost ca. $10, to be collected at door, 2063 Valley Life Sciences Bldg., UC Berkeley campus.
7:00 – talk followed by discussion, in same room.
Reservations required for beverages and dinner (but not the talk). Please email reservations to your host, Brent Mishler, at by Sunday, Feb. 6th
For a map of campus and view of VLSB, use the link below.
This paper, by Clément Gilbert, Cédric Feschotte is quite cool. In it they describe their work on “Paleovirology” where they look for viruses than have “endogenized” by inserting into the genome of some host species. This endogenization is important in particular when the endogenous form becomes inactive and thus, in essence, trapped in the genome. This in turn is important because many viruses evolve so rapidly when they are “free” that it is very hard to reconstruct their ancient history through comparative analysis. But the endogenized viruses serve in essence as a molecular “fossil record” that aids in the comparison and phylogenetic analysis of various families of viruses. As we get more and more genomes, this searching for and analysis of endogenous viruses will get much better.
Anyway, in the paper they report on endogenous viruses in the Zebra Finch genome that are in the Hepadnaviridae family. Here is their summary:
Paleovirology is the study of ancient viruses and the way they have shaped the innate immune system of their hosts over millions of years. One way to reconstruct the deep evolution of viruses is to search for viral sequences “fossilized” at different evolutionary time points in the genome of their hosts. Besides retroviruses, few virus families are known to have deposited molecular relics in their host’s genomes. Here we report on the discovery of multiple fragments of viruses belonging to the Hepadnaviridae family (which includes the human hepatitis B viruses) fossilized in the genome of the zebra finch. We show that some of these fragments infiltrated the germline genome of passerine birds more than 19 million years ago, which implies that hepadnaviruses are much older than previously thought. Based on this age, we can infer a long-term avian hepadnavirus substitution rate, which is a 1,000-fold slower than all short-term substitution rates calculated based on extant hepadnavirus sequences. These results call for a reevaluation of the long-term evolution of Hepadnaviridae, and indicate that some exogenous hepadnaviruses may still be circulating today in various passerine birds.
Figure 4. Summary of the evolutionary scenario inferred in this study.
It is an interesting paper and worth a look if for those who have any interest in viral evolution. And I am becoming more and more fascinated by “Paleovirology” these days so I thought I would just post about this article here. And I guess I am not alone in this opinion that the article is interesting (though I am late). Here is some coverage of their paper:
The paper is actually pretty interesting. But the last line of the abstract. OMG. It is beyond awful. Here is the full abstract:
The fossil record demonstrates that each major taxonomic group has a consistent net rate of diversification and a limit to its species richness. It has been thought that long-term changes in the dominance of major taxonomic groups can be predicted from these characteristics. However, new analyses show that diversity limits may rise or fall in response to adaptive radiations or extinctions. These changes are idiosyncratic and occur at different times in each taxa. For example, the end-Permian mass extinction permanently reduced the diversity of important, previously dominant groups such as brachiopods and crinoids. The current global crisis may therefore permanently alter the biosphere’s taxonomic composition by changing the rules of evolution.
That last line saying that the current extinction crisis may change the rules of evolution really really really bugs me. Changing the rules? Please. If they are rules, then, just how, exactly do they change? If they do change, perhaps they should not be rules no?
And as an aside, what is up with Science not printing the full first name of authors? Does that really save space?
Anyway – not much to say here other than that J. Alroy is the winner of my the 8th “Twisted Tree of Life Award” for suggesting that the evidence presented in this Science paper changes the rules of evolution. And a half award goes to the editors of Science for letting this BS get into the abstract.































































































{kind=link}