—————————————
I first want to than Jonathan for giving me this opportunity. I am a big fan of “behind the science” stories, a habit I fed in grad school by reading every Perspectives (from the journal Genetics) article that I could get a hold of. Science can be rough, but I remember finding solace in stories about the false starts and triumphs of other researchers and how randomness and luck manage to figure into any discovery. If anything I hope to use this space to document this as it is fresh in my mind so that (inevitably) when the bad science days roll around I can have something to look back on. In the very least, I’m looking forward to mining this space in the future for quotes to prove just how little I truly understood about my research topics in 2011. It took a village to get this paper published, so apologies in advance to those that I fail to mention. Also want to mention this upfront,
Marc Nishimura is my co-author and had a hand in every single aspect of this paper.
Joining the Dangl Lab
This project really started way back in 2006, when I interviewed for a postdoc with
Jeff Dangl at UNC Chapel Hill. In grad school I had focused on understanding microbial evolution and genetics but I figured that the best use of my postdoc would be to learn and understand genomics and bioinformatics. I was just about to finish up my PhD and was lucky enough to have some choices when it came around to choosing what to do next. I actually had no clue about Dangl’s research until stumbling across one of his papers in Genetics, which gave me the impression that he was interested in bringing an evolutionary approach to studies of the plant pathogen
Pseudomonas syringae. I was interested in plant pathogens because, while I wanted to study host/pathogen evolution, my grad school projects on
Helicobacter pylori showed me just how much fun it is dealing with the bureaucracy of handling human pathogens. There is extensive overlap in the mechanisms of pathogenesis between plant and human pathogens, but no one really cares how many Arabidopsis plants you infect or if you dispose of them humanely (so long as the transgenes remain out of nature!). By the time I interviewed with Jeff I was leaning towards joining a different lab, but the visit to Chapel Hill went very well and by the end I was primed for Dangl’s sales pitch. This went something along the lines of “look, you can go join another lab and do excellent work that would be the same kinds of things that you did in grad school…or you can come here and be challenged by jumping into the unknown”. How can you turn that down? Jeff sold me on continuing a project started by
Jeff Chang (now a PI at Oregon State), on categorizing the diversity of virulence proteins (type III effector proteins to be exact) that were translocated into hosts by the plant pathogen P
seudomonas syringae. Type III effectors are one of the main determinants of virulence in numerous gram negative plant and animal pathogens and are translocated into host cells to ultimately disrupt immune functions (I’m simplifying a lot here). Chang had already created genomic libraries and had screened through random genomic fragments of numerous
P. syringae genomes to identify all of the type III effectors within 8 or so phylogenetically diverse strains. The hope was that they would find a bunch of new effectors by screening strains from different hosts. Although this method worked well for IDing potential effectors, I was under the impression that it was going to be difficult to place and verify these effectors without more genomic information. I was therefore brought in to figure out a way to sequence numerous
P. syringae genomes without burning through a Scrooge McDuckian money bin worth of grant money. We had a thought that some type of grand pattern would emerge after pooling all this data but really we were taking a shot in the dark.

Tomato leaves after 10 days infection by the tomato pathogen P.syringae DC3000 (left) as well as a less virulent strain (right). Disease symptoms are dependent on a type III secretion system.
Moments of Randomness that Shape Science
When I actually started the postdoc, next generation sequencing technologies were just beginning to take off. It was becoming routine to use 454 sequencing to generate bacterial genome sequences, although Sanger sequencing was still necessary to close these genomes. Dangl had it in his mind that there had to be a way to capitalize on the developing Solexa (later Illumina) technology in order to sequence
P. syringae genomes. There were a couple of strokes of luck here that conspired to make this project completely worthwhile. I arrived at UNC about a year before the UNC Genome Analysis core facility came online. Sequencing runs during the early years of this core facility were subsidized by UNC, so we were able to sequence many Illumina libraries very cheaply. This gave us the opportunity to play around with sequencing options at low cost, so we could explore parameter space and find the best sequencing strategy. This also meant that I was able to learn the ins and outs of making libraries at the same time as those working in the core facility (Piotr Mieczkowski was a tremendous resource). Secondly, I started this postdoc without knowing a lick of UNIX or perl and knew that I was going to have to learn these if I had any hope of assembling and analyzing genomes. I was very lucky to have
Corbin Jones and his lab 3 floors above me in the same building to help work through my kindergarden level programming skills. Corbin was really instrumental to all of these projects as well as in keeping me sane and I doubt that these projects would have turned out anywhere near as well without him. Lastly, plant pathogens in general, and
P. syringae in particular, were poised to greatly benefit from next generation sequencing in 2006. While there was ample funding to completely sequence (close) genomes for numerous human pathogens, lower funding opportunities for plant pathogens meant that we were forced to be more creative if we were going to pull of sequencing a variety of P.syringae strains. This pushed us into trying a NGS approach in the first place. I suspect that it’s no coincidence that, independently of our group, the NGS assembler
Velvet was first utilized for assembling
P.syringae isolates.
The Frustrations of Library Making
Through a collaboration with
Elaine Mardis’s group at Washington University St. Louis, we got some initial data back that suggested it would be difficult to make sense of bacterial genomes at that time using only Illumina (the paired end kits weren’t released until later). There simply wasn’t good enough coverage of the genome to create quality assemblies with the assemblers available at this time (
SSAKE and
VCAKE, our own (really Will Jeck’s) take on SSAKE). Therefore we decided to try a hybrid approach, combining low coverage 454 runs (initially separate GS Flex runs with regular reads and paired ends, and later one run with long paired ends) with Illumina reads to fill in the gaps and leveraging this data to correct for any biases inherent in the different sequencing technologies. Since there was no core facility at UNC when I started making libraries, I had to travel around in order to find the necessary equipment. The closest place that I could find a machine to precisely shear DNA was
Fred Dietrich’s lab at Duke. More than a handful of mornings were spent riding a TTA bus from UNC to Duke, with a cooler full of genomic DNA on dry ice (most times having to explain to the bus drivers how I wasn’t hauling anything dangerous), spending a couple of hours on Fred’s hydroshear, then returning to UNC hoping that everything worked well. There really is no feeling like spending a half a day travelling/shearing only to find out that the genomic DNA ended up the wrong size. We were actually planning to sequence one more strain of
P. syringae, and already had Illumina data, but left this one out because we filled two plates of 454 sequencing and didn’t have room for a ninth strain. In the end there were two very closely related strains (
P.syringae aptata or
P. syringae atrofaciens) left to make libraries for and the aptata genome sheared better on the last trip than atrofaciens. If you’ve ever wondered why researchers pick certain strains to analyze, know that sometimes it just comes down to which strain worked first. Sometimes there were problems even when the DNA was processed correctly. I initially had trouble making the 454 libraries correctly in that, although I would follow the protocol exactly, I would lose the DNA somewhere before the final step. I was able to trace down the problem to using an old (I have no clue when the Dangl lab bought it, but it looked as useable as salmon sperm ever does) bottle of salmon sperm DNA during library prep. There were also a couple of times that I successfully constructed Illumina libraries only to have the sequencing runs dominated by few actual sequences. These problems ultimately stemmed from trying to use homebrew kits (I think) for constructing Illumina libraries. Once these problems were resolved,
Josie Reinhardt managed to pull everything together and create a pipeline for hybrid genome assembly and we published our first hybrid genome assembly in Genome Research. At that moment it was a thrill that we could actually assemble a genome for such a low cost. It definitely wasn’t a completely sequenced genome, but it was enough to make calls about the presence or absence of genes.
Waiting for the story to Emerge
There are multiple ways to perform research. We are all taught about how important it is to define testable hypothesis and to set up appropriate experiments to falsify these educated guesses. Lately, thanks to the age of genomics, it has become easier and feasible to accumulate as much genomic data as possible and find stories within that data. We took this approach with the Pseudomonas syringae genome sequences because we knew that there was going to be a wealth of information, and it was just a matter of what to focus on. Starting my postdoc I was optimistic that our sampling scheme would allow us to test questions about how host range evolves within plant pathogens (and conversely, identify the genes that control host range) because the strains we were going to sequence were all isolated from a variety of diseased hosts. My naive viewpoint was that we were going to be able to categorize virulence genes across all these strains, compare suites of virulence genes from strains that were pathogens of different hosts, and voila…we would understand host range evolution. The more I started reading about plant pathology the more I became convinced that this approach was limited. The biggest problem is that, unlike some pathogens, P. syringae can persist in a variety of environments with strains able to survive our flourish or on a variety of hosts. Sure we had strains that were known pathogens of certain host plants, but you can’t just assume that these are the only relevant hosts. Subjective definitions are not your friend when wading into the waters of genomic comparisons.
We were quite surprised that, although type III effectors are gained and lost rapidly across
P.syringae and our sequenced strains were isolated from diverse hosts, we only managed to identify a handful of new effector families. I should also mention here that
Artur Romanchuk came on board and did an extensive amount of work analyzing gene repertoires across strains. A couple of nice stories did ultimately emerge by comparing gene sequences across strains and matching these up with virulence in planta (we are able to show how mutation and recombination altered two different virulence genes across strains), but my two favorite stories from this paper came about from my habit of persistently staring at genome sequences and annotations. As I said above, a major goal of this paper was to categorize the suites of a particular type of virulence gene (type III effectors) across
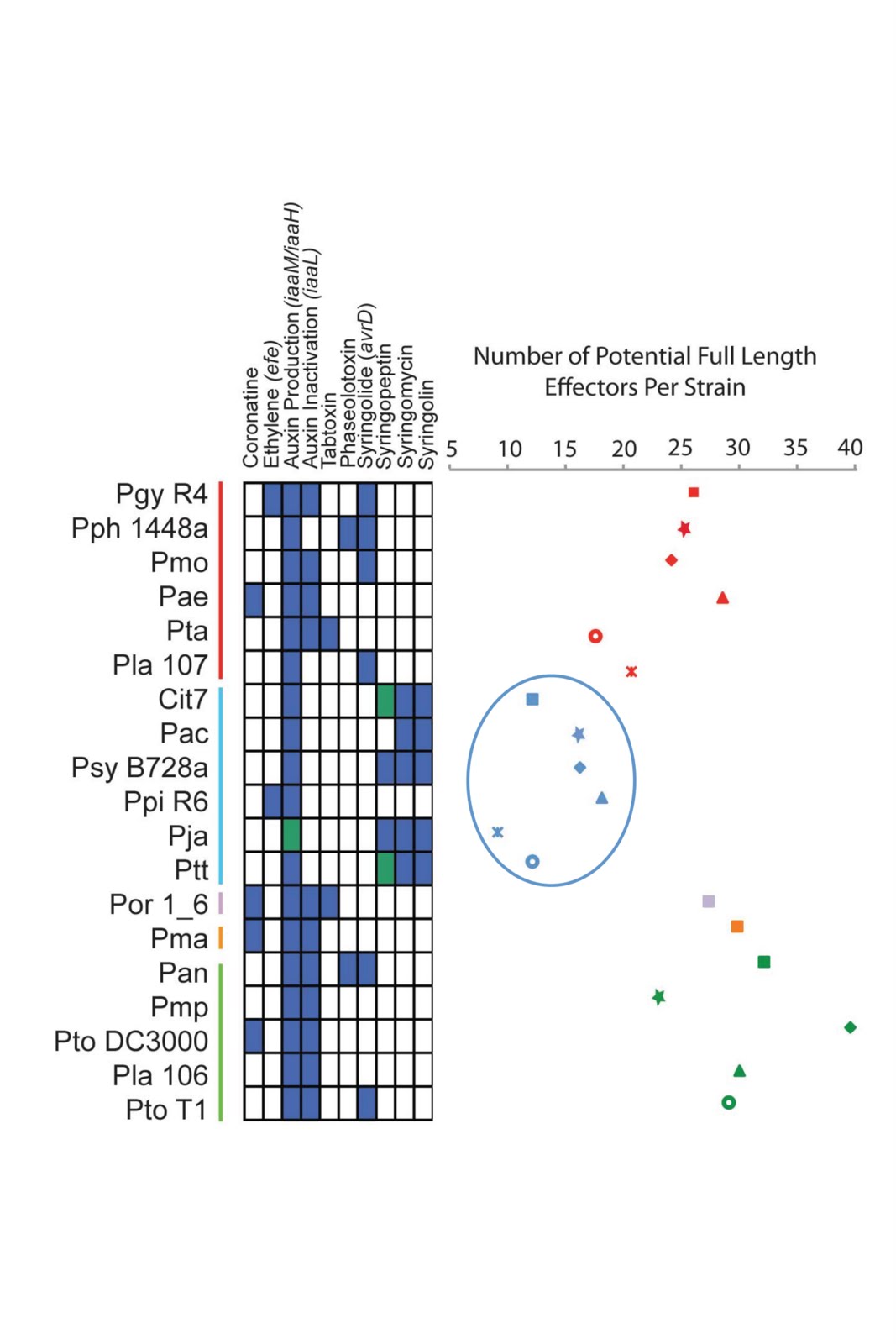
P. syringae. I was staring at gene repertoires across strains when I noticed that two of the strains had very few of these effectors (10 or so) compared to most of the other strains (20-30). When I plotted total numbers of effectors across strains, a phylogenetic pattern arose where genomes from a subset of closely related
P. syringae strains possessed lower numbers of effectors. I then got the idea to survey for other classes of virulence genes, and sure enough, strains with the lowest numbers of effectors all shared pathways for the production of well characterized toxin genes (Non ribosomal peptide synthase (NRPS) toxins are secreted out of
P. syringae cells and are virulence factors, but are not translocated through the type III secretion system). One exception did arise across this handful of strains (a pea pathogen isolate from pathovar pisi) in that this strain has lost each of these conserved toxin pathways and also contain the highest number of effectors within this phylogenetic group. The relationship between effector number and toxin presence remains a correlation at the present time, but I’m excited to be able to try and figure out what this means in my own lab.

Modified Figure 3 from the paper. Strain names are listed on the left and are color coded for phylogenetic similarity. Blue boxes indicate that the virulence gene/toxin pathway is present, green indicates that the pathway is likely present but sequence was truncated or incomplete, while box indicates absence. I have circled the group II strains, which have the lowest numbers of type III effectors while also having two conserved toxin pathways (syringomycin and syringolin). Note that the Pisi strain (Ppi R6) lacks these toxin pathways.
The other story was a complete stroke of luck. P. syringae genomes are typically 6Mb (6 million base pairs) in size, but one strain that we sequenced (a cucumber pathogen) contained an extra 1Mb of sequence. Moreoever, the two largest assembled contigs from this strain were full of genes that weren’t present in any other P. syringae strain. After some similarity comparisons, I learned that there was a small bit of overlap between each of these contigs and performed PCR to confirm this. Then, as a hunch, I designed primers facing out of each end of the contig and was able to confirm that this extra 1Mb of sequence was circular in conformation and likely separate from the chromosome. I got a bit lucky here because there was a small bit (500bp or so) of sequence that was not assembled with either of these two contigs that closed the circle (a lot more and I wouldn’t have gotten the PCR to work at all). We quickly obtained 3 other closely related strains and were able to show that only a subset of strains contain this extra 1Mb and that it doesn’t appear to be directly involved in virulence on cucumber. So it turns out that a small number (2 so far) of P. syringae strains have acquired and extra 1Mb of DNA, and we don’t quite know what any of these ~700 extra genes do. There are no obvious pathways present aside from additional chromosomal maintenance genes, extra tRNAs in the same ratio as the chromosomal copies, and a couple of secretion systems. So somehow we managed to randomly pick the right strain to capture a very recent event that increased the genome size of this one strain by 15% or so. We’ve made some headway on this megaplasmid story since I started my lab, but I’ll save that for future blog posts.

Modified Figure S12 from the paper. Strains that contain the 1Mb megaplasmid (Pla7512 and Pla107) are slightly less virulent during growth in cucumber than strains lacking the megaplasmid (PlaYM8003, PlaYM7902). This growth defect is also measurable in vitro. In case you are wondering, I used blue and yellow because those were the dolors of my undergrad university, the University of Delaware.Reviewer Critiques
We finally managed to get this manuscript written up by the summer of 2010 and submitted it to PLoS Biology. I figured that (as always) it would take a bit of work to address reviewer’s critiques, but we would nonetheless be able to publish without great difficulty. I was at a conference on P. syringae at Oxford in August of 2010 when I got the reviews back and learned that our paper had gotten rejected. Everyone has stories about reviewer comments and so I’d like to share one of my own favorites thus far. I don’t think it ever gets easier to read reviews when your paper has been rejected, but I was knocked back the main critique of one reviewer:
“I realize that the investigators might not typically work in the field of bacterial genomics, but when looking at divergent strains (as opposed to resequencing to uncover SNPs among strains) it is really necessary to have complete, not draft, genomes. I realize that this might sound like a lot to ask, but if they look at comparisons of, for example, bacterial core and pan-genomes, such as the other paper on this that they cite (and numerous other examples exist), they are based on complete genome sequences. If this group does not wish to come up to the standards applied to even the most conventional bacterial genomics paper, it is their prerogative; however, they should be aware of the expectations of researchers in this field.”
So this reviewer was basically asking us to spend an extra 50k to finish the genomes for these strains before they were scientifically useful. Although I do understand the point, this paper was never about getting things perfect but about demonstrating what is possible with draft genomes. I took the part about working in the field of bacterial genomics a bit personally I have to admit, c’mon that’s harsh, but I got over that feeling by downing a few pints in Oxford with other researchers that (judging by their research and interest in NGS) also failed to grasp the importance of spending time and money to close P. syringae genomes. We managed to rewrite this paper to address most of the other reviewers critiques and finally were able to submit to PLoS Pathogens.
Baltrus DA, Nishimura TM, Reinhardt JA, Romanchuk A, Chang JH, Mukhtar MS, Cherkis K, Roach J, Grant SR, Jones CD, Dangl JL “Dynamic evolution of pathogenicity revealed by sequencing and comparative genomics of 19 Pseudomonas syringae isolates” PLoS Pathogens 7(7):e1002132