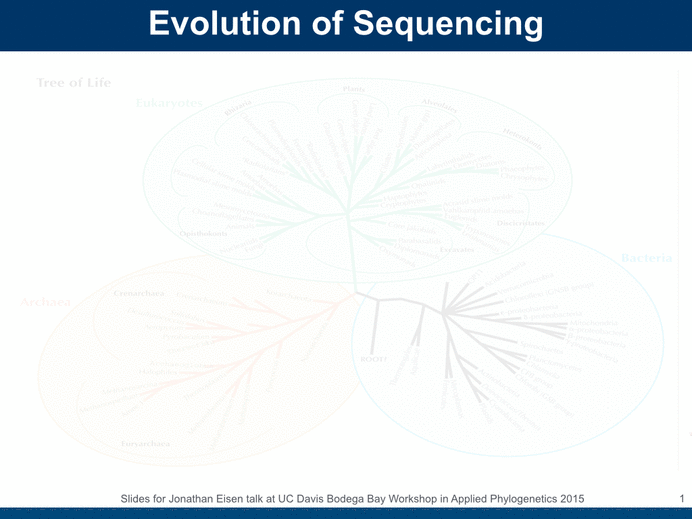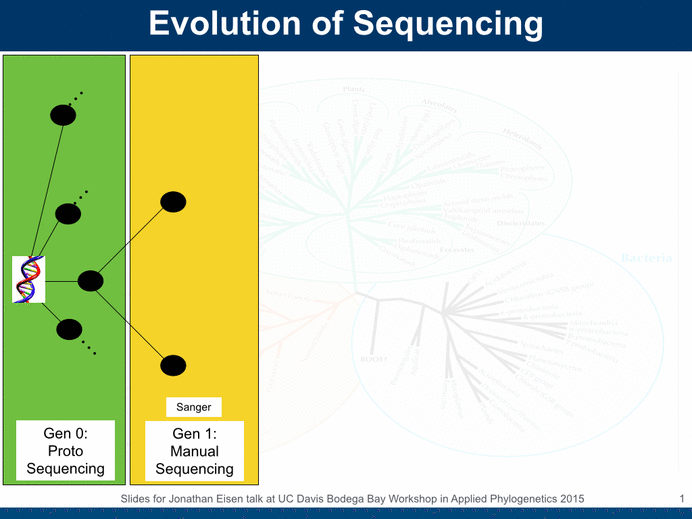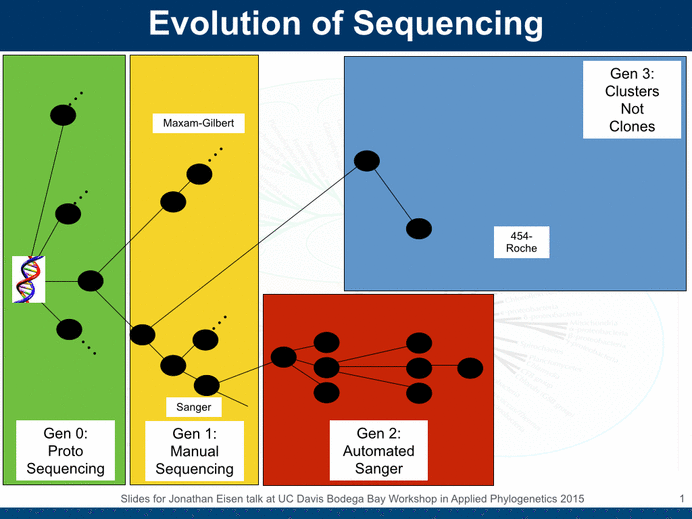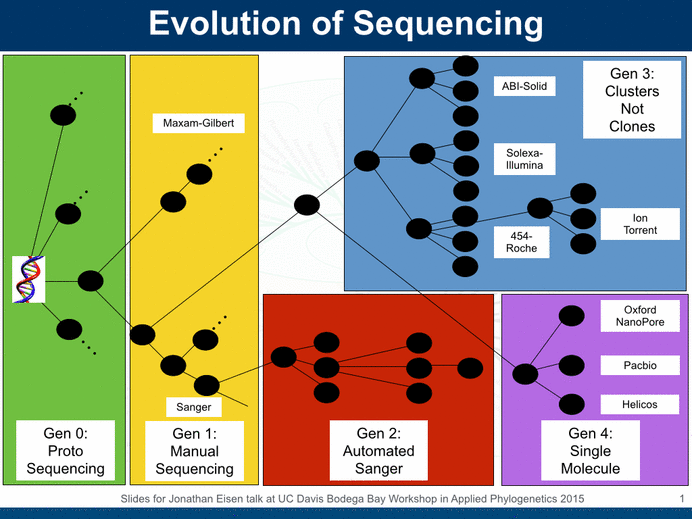
For the last year or so I have become a big fan of Illumina sequencing. We are using it for everything in the lab. And many others are using it quite a lot too. All sorts of interesting applications. But of course -there are other sequencing systems that each have some advantages relative to Illumina. And one of the key limitations of Illumina sequencing has been the read length (though that limitation gets less and less as read lengths get longer and longer from Illumina machines).
The UC Davis Genome Center has had Illumina sequencing systems for many years now and we use them extensively. However, we felt for some time that we and others around town could benefit from complementary methods, especially those that could get longer reads. So we sought funding to buy other systems. And fortunately we got an NSF MRI grant to do just that -which we used to buy a Roche 454 Jr machine and contribute to the purchase of a Pacific Biosciences machine. These are good to have around because they open up new windows into sequencing – not just long reads but other areas as well. For example, the PacBio system also has the ability to use it to detect modifications to bases like methylation.
Alas, both the 454 and PacBio systems have higher error rates than the Illumina systems. And this makes some analyses challenging and limits the benefits that come from the longer reads. So what to do? For a while people have been using Illumina sequencing to “correct” the errors make by 454 and PacBio sequencing. And today Matt Herper at Forbes (For A New DNA Sequencer, A Technical Fix May Have Come Too Late – Forbes) discusses a new further improvement in the ability to do this error correction (a paper just came out on the topic from Adam Phillippy, Sergey Koren, Michael Schatz, and others).
I find this whole concept a bit funny / interesting. Not only does Illumina sequencing have many uses but one of its uses in essence helps keep aloft the potential of some of it’s competitors. In this way – Illumina can be considered the duct tape of sequencing systems. 1001 uses. Not sure the Illumina folks will be overly thrilled with this use but that is the way it goes …
(As an aside – any high throughput highly accurate sequencing method could be used in the same way as Illumina in most cases – ABI solid for example. But alas for ABI Illumina has kind of taken over this part of the market).
(An another aside – we will have to wait and see how/if the Ion Torrent systems take off in the sequencing ecosystem)
(As another aside – still waiting to see some more detail from the Oxford Nanopores folks … I would be happy to be a beta tester if anyone from Oxford is reading this).





{kind=link}
{kind=link}