This is another in my ongoing “Story behind the paper series”. This one is from Christophe Dessimoz on a new paper he is an author on in PLoS Computational Biology that is near and dear to my heart.
See below for more. I am trying to post this from Yosemite National Park without full computer access so I hope the images come through. If not I will fix in a few days.
……………..
I’d like to thank Jonathan for the opportunity to tell the story behind our paper, which was just published in PLoS Computational Biology. In this work, we corroborated the “ortholog conjecture”—the widespread but little tested notion that orthologs tend to be functionally more conserved than paralogs.
I’d also like to explore more general issues, including the pitfalls of statistical analyses on highly heterogeneous data such as the Gene Ontology, and the pivotal role of peer-reviewers.
Like many others in computational biology, this project started as a quick analysis that was meant to take “just a few hours” but ended up keeping us busy for several years…
The ortholog conjecture and alternative hypotheses
The ortholog conjecture states that on average and for similar levels of sequence divergence, genes that started diverging through speciation (“orthologs”) are more similar in function than genes that started diverging through duplication (“paralogs”). This is based on the idea that gene duplication is a driving force behind function innovation. Intuitively, this makes sense because the extra copy arising through duplication should provide the freedom to evolve new function. This is the conventional dogma.
Alternatively, for similar levels of sequence divergence, there might not be any particular difference between orthologs and paralogs. It is the simplest explanation (per Ockham’s razor), and it also makes sense if the function of a gene is mainly determined by its protein sequence (let’s just consider one product per gene). Following this hypothesis, we might expect considerable correlation between sequence and function similarity.
But these are by no means the only two possible hypotheses. Notably, Nehrt and colleagues saw higher function conservation among within-species homologs than between-species homologs, which prompted them to conclude: “the most important aspect of functional similarity is not sequence similarity, but rather contextual similarity”. If the environment (“the context”) is indeed the primary evolutionary driving force, it is not unreasonable to speculate that within-species paralogs could evolve in a correlated manner, and thus be functionally more similar than their between-species counterparts.
Why bother testing these hypotheses?
Testing these hypotheses is important not only for better understanding gene function evolution in general, but it also has practical implications. The vast majority of gene function annotations (98% of Gene Ontology annotations) are propagated computationally from experimental data on a handful of model organisms, often using models based on these hypotheses.
How our work started
Our project was born during a break at the 10th anniversary meeting of the Swiss Institute of Bioinformatics in September 2008. I was telling Marc Robinson-Rechavi (University of Lausanne) about my work with Adrian Altenhoff on orthology benchmarking (as it happens, another paper edited by Jonathan!), which had used function similarity as a surrogate measure for orthology. We had implicitly assumed that the ortholog conjecture was true—a fact that Marc zeroed in on. He was quite sceptical of the ortholog conjecture, and around this time, together with his graduate student Romain Studer, he published an opinion in Trends in Genetics unambiguously entitled “How confident can we be that orthologs are similar, but paralogs differ?” (self-archived preprint). So, having all that data on hand, we flipped our analysis on its head and set out to compare the average Gene Ontology (GO) annotation similarity of orthologs vs. paralogs. Little did we think that this analysis would keep us busy for over 3 years!
First attempt
It only took a few weeks to obtain our first results. But we were very puzzled. As Nehrt et al. would later publish, we observed that within-species paralogs tended to be functionally more conserved than orthologs. At first we were very sceptical. After all, Marc had been leaning toward the uniform ortholog/paralog hypothesis, and I had expected the ortholog conjecture to hold. We started controlling for all sorts of potential sources of biases and structure in the data (e.g. source of ortholog/paralog predictions, function and sequence similarity measures, variation among species clades). A year into the project, our supplementary materials had grown to a 67-page PDF chock-full of plots! The initial observation held under all conditions. By then, we were starting to feel that our results were not artefactual and that it was time to communicate our results. (We were also running out of ideas for additional controls and were hoping that peer-reviewers might help!)
Rejections
We tried to publish the paper in a top-tier journal, but our manuscript was rejected prior to peer-review. I found it frustrating that although the work was deemed important, they rejected it prior to review over an alleged technical deficiency. In my view, technical assessments should be deferred to the peer-review process, when referees have the time to scrutinise the details of a manuscript.
Genome Research sent our manuscript out for peer-review, and we received one critical, but insightful report. The referee contended that our results were due to species-specific factors, which arise because “paralogs in the same species tend to be ‘handled’ together, by experimenters and annotators”. The argument built on one example which we had discussed in the paper: S. cerevisiae Cdc10/Cdc12 and S. pombe Spn2/Spn4 are paralogs inside each species, while Cdc10/Spn2 and Cdc12/Spn4 are the respective pairs of orthologs. The Gene Ontology annotations for the orthologs were very different, while the annotations of theparalogs were very similar. The reviewer looked at the source articles indetail, and noticed that “the functional divergence between these genes is more apparent than real”. Both pairs of paralogs were components of theseptin ring. The differences in annotation appeared to be due to differences in the experiments done and in the way they were transcribed. The reviewer stated:
“A single paper will often examine the phenotype of several paralogs within onespecies, resulting in one paper, which is presumably processed by one GO annotator at one time. In contrast, phenotypes of orthologs in different species almost always come from different papers, via different annotation teams.”
Authorship effect: an easily overlooked bias
At first, it was tempting to just dismiss the criticism. After all, as Roger Brinner put it, “the plural of anecdote is not data.” More importantly, we had tried to address several potential species-specific biases, such as uneven annotation frequencies among species (e.g. due to developmental genes being predominantly studied in C. elegans). And we had been cautious in our conclusions, suggesting that our results might be due to an as yet unknown confounder in the Gene Ontology dataset (remember that we had run out of ideas?). So the referee was not telling us anything we did not know.
Or was (s)he? Stimulated by the metaphor of same-species paralogs being “handled” together, we decided to investigate whether common authorship might correlate in any way with function annotation similarity. Here’s what we observed:
The similarity of function annotations from a common paper is much higher than otherwise! Even if we restrict ourselves to annotations from different papers, but with at least one author in common, the similarity of functional annotations is still considerably higher than with papers without a common author.
Simpson’s Paradox
In itself, the authorship effect is not necessarily a problem: if annotations between orthologs and paralogs were similarly distributed among the different types, differences due to authorship effects would average out. The problem here is that paralogs are one order of magnitude more likely to be annotated by the same lab than orthologs. This gives rise to “Simpson’s paradox”: paralogs can appear to be functionally more similar than orthologs just because paralogs are much more likely to be studied by the same people.
A classical example of Simpson’s paradox is the “Berkeley gender bias case” (Wikipedia article): the university was sued for bias against women applicants based on the aggregate 1973 admission figures (44% men admitted vs. 35% women). As it turned out, the admission rate for each department was in fact similar for both sexes (and even in favour of women in a few departments). The lower overall acceptance rate for women was not due to gender bias, but to the tendency of women to apply to more competitive departments.
Paper by Nehrt et al.
Finding the authorship effect meant that we had to reanalyse all our data, and completely rewrite our manuscript. A few months into this process, in June 2011, Matt Hahn and colleagues published their paper (Nehrt et al., Testing the Ortholog Conjecture with Comparative Functional Genomic Data from Mammals, PLoS Comput Biol 2011). Matt has written a very interesting story behind the paper on this blog, which is well-worth reading (including comments).
While we weren’t very surprised by the essence of their observations—they were very consistent with our initial (rejected) manuscript—we were nevertheless struck by the similarity in the presentation of the results:
On the left, plot 2A from Nehrt et al., PLoS Comput Biol 2011; On the right, plot from our initial, rejected manuscript. Note that their blue (within-spec outparalogs) and green (inparalogs) lines are combined in our plot (same-species paralogs, yellow line)
The publication of Nehrt et al.’s gave us mixed feelings. Obviously, their work was taking away some of the novelty in our study. But at the same time, they were drawing considerable attention to the problem (not least by coining a catchy name to describe the question!). And of course, we already knew at this point that their observations were confounded by factors such as the authorship effect, so it was not the end of the story.
Is it possible to draw reliable conclusions from observational data such as GO annotations?
Before I move to our findings, it’s worth pondering a bit more on the issue of biases in the data. Statisticians and epidemiologists make a strong distinction between experimental data (=data from a controlled experiment, designed such that the case and control groups are as identical as possible in all respects other than a factor of interest) and observational data (=data lying around). (On a side note, Ewan Birney recently wrote a great post on study design in genetic and phenotypic studies, with several ideas relevant to this issue.) Data pulled from the GO database clearly falls into the second category: observational data. We are at the mercy of potentially countless hidden effect biasing our conclusions in all sorts of ways.
Can we rely on this data at all? For some, the answer appears to be a categorical “no“. A more pragmatic stance was expressed by the GO consortium in a recent reply to Nehrt et al.’s paper, which identified potential confounding effects ignored in the study of Nehrt et al., such as species-specific annotation biases (they suggested, tongue-in-cheek, that the study instead supports the “biased annotation conjecture”), and stressed that “users of GO should ensure that they test for, and adjust for, potential biases prior to interpretation”.
In the end, I think that this debate, consistent with our experience, highlights the risks of working with observational data. But at the same time, observational data is often all we have (or can afford), so the best course of action seems indeed to control for known confounders, try to identify unknown ones, and cautiously process forward.
Resolving the conjecture
Controlling for the authorship effect and other biases—some previously known, others newly identified—we found that for similar levels of divergence, orthologs tend to be functionally more conserved than paralogs. This is true for different source of orthology/paralogy predictions, different types of function and sequence similarity measures, and different data sampling strategies. But in absolute terms, the difference is often not very strong, and varies considerably among species and types of functions. Our study confirms the ortholog conjecture, but at the same time it shows that the conjecture is not very useful in practice as it does not have much predictive power.
They were two crucial contributors to this study: the peer-reviewers, and open science. We are obviously indebted to the reviewer who rejected our paper on the basis of a potential authorship bias. The reviewers of the resubmission provided detailed and highly competent feedback As for open science, where would computational biology be without it? We often take it for granted, but without publicly available genome data and functional annotations, a study like this would never have happened. Chemistry suffers from having only very few publicly available databases (e.g. ChEMBL). People hard at work behind model organism databases have our deepest appreciation.
Upcoming discussions
As far as I can tell, Nehrt et al.’s study caused a considerable stir in our community, but many critics could not quite put their finger precisely on what was wrong (see Matt Hahn’s post, especially section “The fallout, and some responses”). Our work explains and reconciles their controversial observations.
This is by no means the end of the discussion. There will be a symposium dedicated to the ortholog conjecture at SMBE in Dublin next month (let me know if you want to meet up), and it will almost certainly be a topic of the next Quest for Orthologs meeting (tentatively scheduled for the summer 2013). Meanwhile, I hope this work will spur discussions on this blog (or, in French, on Marc Robinson-Rechavi’s blog!) and/or on Twitter (you can follow me at @cdessimoz, and Marc at @marc_rr).
Thanks to Marc Robinson-Rechavi and Mary Todd Bergman for their feedback on drafts of this post.






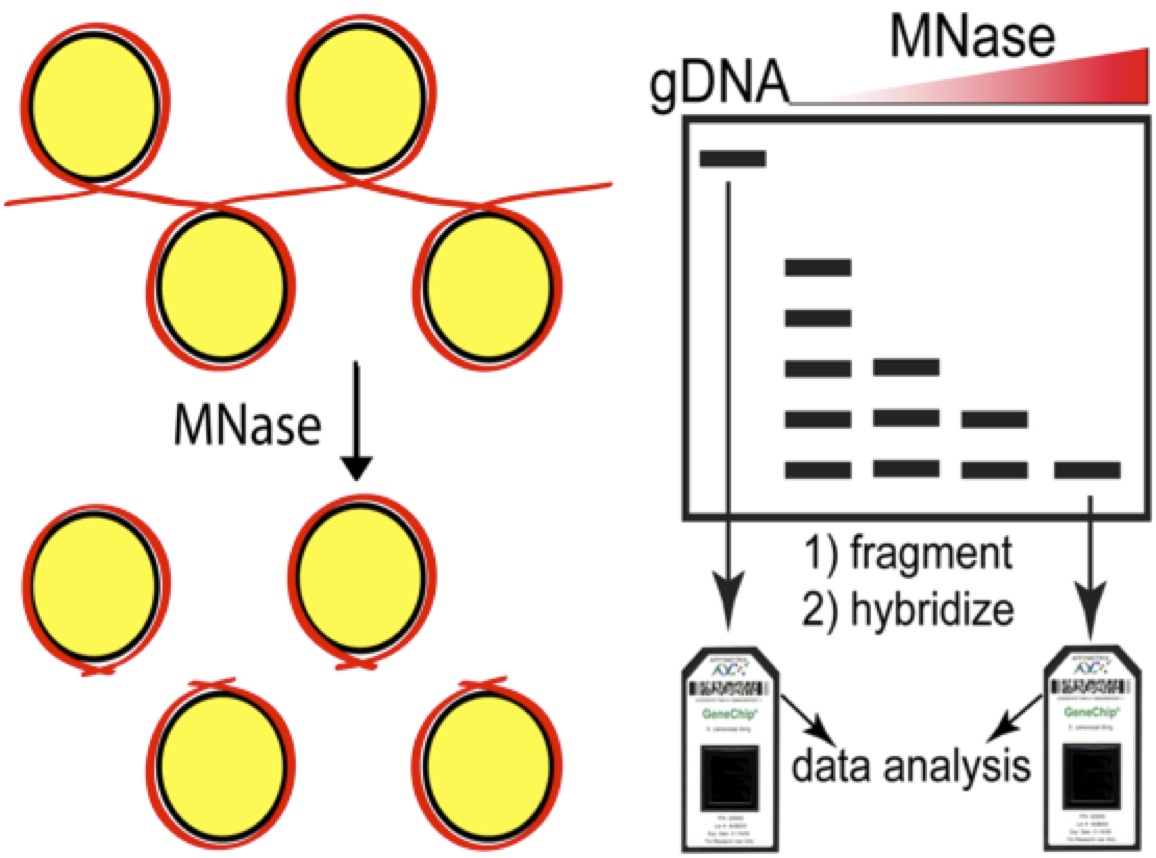




 This was a pleasant surprise and one we did not take for granted given the high CG content of the genome (65%). We then turned to isolating RNA. Without polyA tails for enrichment, our first attempts at RNA-seq were 95% ribosomal. Combining partially successful double-stranded nuclease (DSN) treatment with massive sequencing depth we were able to get fairly high coverage of the transcriptome. Here’s where Ron Ammar, a graduate student supervised by me, Guri Giaever and Gary Bader stepped in and turned my laboratory adventures into a wonderful story. Ron mapped the reads from our nucleosome samples to the reference genome and found what to my eyes looked like a yeast nucleosome map only at half scale.
This was a pleasant surprise and one we did not take for granted given the high CG content of the genome (65%). We then turned to isolating RNA. Without polyA tails for enrichment, our first attempts at RNA-seq were 95% ribosomal. Combining partially successful double-stranded nuclease (DSN) treatment with massive sequencing depth we were able to get fairly high coverage of the transcriptome. Here’s where Ron Ammar, a graduate student supervised by me, Guri Giaever and Gary Bader stepped in and turned my laboratory adventures into a wonderful story. Ron mapped the reads from our nucleosome samples to the reference genome and found what to my eyes looked like a yeast nucleosome map only at half scale.