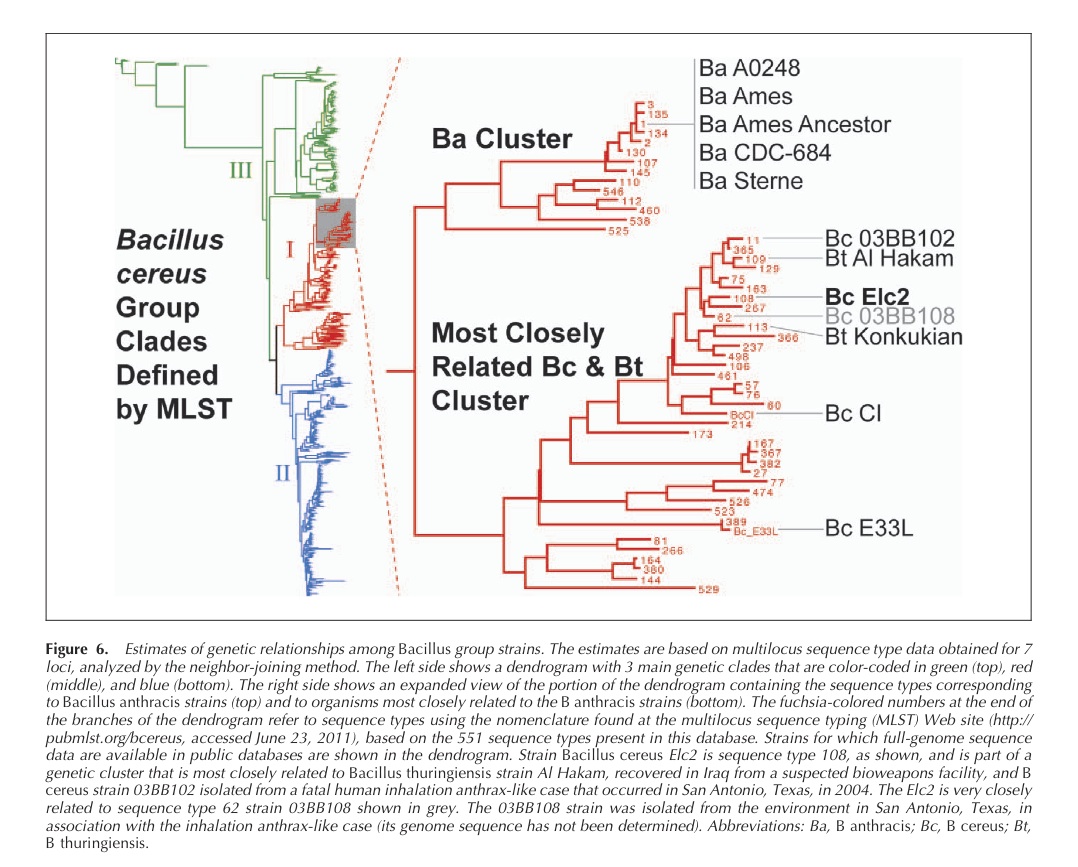
Just got an email marketing a new book on numerology. I have a feeling, just a slight one, the marketer does not know much about me. But I thought I would share so everyone can ponder numerology. In order to spare people if they are not interested I have put everything below the “jump break”
Dear Jonathan Eisen:
Glynis McCants has studied Numerology for over 21 years and her accuracy at predicting events has earned her repeat appearances on The Tyra Banks Show, The Today Show, The View and many others. When she does live, quick Numerology readings on air, SHE DOES NOT MISS! In fact, the producers of Jim Carrey’s movie Number 23 hired her as their expert as did the producers of Dancing with the Stars. Upon its release, her latest book Love by the Numbers became the best-selling Numerology book in the country. Love by the Numbers has received rave reviews for its accuracy in helping single people find lasting love, and giving people in a relationship the tools they need to re ignite the passionate love they once had.
In a radio interview at the beginning of this year, Glynis said that in the month of August, there would be a lot of political upheaval and serious issues with the economy. The recent downgrade of The United States Credit Rating is a great example of this, and Glynis can give more insights on this subject.
Please read the following press release and let me know if I may book an informative, as well as entertaining interview for you with Glynis McCants. Thank you.
Please view Glynis in television appearances at: http://vids.myspace.com/index.cfm?fuseaction=vids.individual&VideoID=23012149
Rick Stevens
Ascot Media Group
Post Office Box 133032
The Woodlands, TX 77393
Office: (281) 333-3507
pr@ascotmediagroup.com
http://www.ascotmedia.com
FOR IMMEDIATE RELEASE
Welcome To Glynis McCants’ World – The World Of Accurately Predicting Romantic Love!
San Marino, CA, August 10, 2011 – Glynis McCants is an internationally-known celebrity Numerologist, whose latest book Love by the Numbers (Sourcebooks Publishing), was written for anyone looking for a healthy love relationship, or wanting to improve the one they already have. With a background in comedy, McCants is an in-demand motivational speaker on relationships and in her quick, live on-air readings – she does not miss!
Knowledge is power, and by researching Numerology for over 21 years and using the 2,500 year-old Pythagoras Number System, Glynis tells us that when you have someone’s name and date of birth, you can discover who they really are. Celebrity couples are one of Glynis’ specialties. She predicted that Reese Witherspoon/Ryan Phillippe, Halle Berry/Eric Benet, Ryan Reynolds/Scarlett Johansson’s marriages would not go the distance. Not only did Glynis know Jennifer Lopez was not going to walk down the aisle with Ben Affleck, but she was certain that Jennifer’s marriage with Marc Anthony would be a very difficult one, and that they would not be living “happily ever after.” Glynis makes it clear that Numerology is in everything– people, our home, business, and even the city and state in which we live.
Naming your baby by the Numbers really does make a difference. In the news, Pink, Jewel, Ivanka Trump, Kate Hudson, and Victoria Beckham have all recently given birth, and Glynis teaches that giving the baby the right name can ensure compatibility with child and parents. As your guest, she can explain how that works.
Glynis’ book Love by the Numbers is incredibly accurate, informative and entertaining! For many years, Glynis was a Numerology Counselor who taught others how to find healthy love relationships. One day, after a period of dating disappointments, she realized that she needed to follow her own advice and apply the science to finding her own compatible relationship. She used the power of Numerology to find her husband Charlie. They have been together for nine years, and are still madly in love, and feel as if they are on an extended honeymoon.
While other books make it complicated, the author has defined each number in a simple, understandable process. Its 25 exciting chapters teach readers how to find the Six Numbers that make up their Numerology Blueprint, and how to do a chart comparison. You can also discover what turns your partner on- or- off in love. Love by the Numbers will also show you how to pick the perfect wedding date to avoid a wedding day disaster! There are instances where taking the husband’s surname can actually harm the relationship—this book lets you know if that is the case for you.
Glynis McCants has made repeat TV appearances on The Tyra Banks Show, The Today Show, The Other Half, The Leeza Show, The View and Entertainment Tonight. She was the Numerology expert on Dancing with the Stars and for Jim Carrey’s film, Number 23. Glynis has been featured in top magazines and is a recurring guest on George Noory’s Coast to Coast Radio. She is also heard weekly on The Leeza At Night radio show. Glynis was voted Favorite Guest on PBR for three years running.
Please visit the Glynis McCants website at: http://www.numberslady.com for more information.
###
TIP SHEET:
Olympia Dukakis: “What impresses me most about Glynis is her boundless hope and optimism about the world we live in today. She uses the ancient science of Numerology as her guide, but her insight and wisdom are invaluable. Glynis believes that you can achieve all of your dreams, and by the time you finish reading this book, you will too.”
Leeza Gibbons, Talk Show Host: “I’ve seen Glynis work her Numerology magic dozens of times…always with the same result; empowering people to create the best version of themselves by really knowing and understanding who they are.”
Caroline Rhea: “Glynis knows so much about numbers, she makes the Yellow Pages seem obsolete.”
L.A. Confidential magazine: “To put it simply, if cupid and a clairvoyant were to have an affair, Glynis McCants would very much look like their offspring.”
John Edward, Psychic Medium: “When I think about Numerology I think about two people: Pythagoras and his modern day equivalent, Glynis McCants, The Numbers Lady. Glynis has taken a historic metaphysical science and transformed it into an easy to understand, organized and quite honestly entertaining field of study. To sum it up, she’s GOT your number – now it’s time for you to get it.”