Well, today in my “Microbial phylogenomics” class at UC Davis we are discussing what I think might be the most important paper (well, actually, series of papers) in the history of microbiology. The papers are the ones where Carl Woese, George Fox and colleagues outline the evidence for the existence of a “hidden” third major branch in the tree of life – what is now known as the archaea. The evidence for this third branch was first laid out in a series of papers in 1977 including:
Now Woese, Fox and others in Woese’s group had been leading up to these publications in ways for years (I note, there were some pretty incredible people involved in these studies in the years before 1977 too including Mitch Sogin, now at MBL, David Stahl, Chuck Kurland, Norm Pace, etc but that is another story). They had been determining the nucletide sequences of small fragments of rRNAs from different species, especially from different organisms that did not have nuclei – the so-called “prokaryotes”. And they were using these sequences to infer the phylogenetic relationships among these microbes.
Consider for example, the paper by SJ Sogin et al in 1972 “
Phylogenetic measurement in procaryotes by primary structural characterization. Sogin SJ, Sogin ML, Woese CR.
J Mol Evol. 1971;1(1):173-84. This paper laid out some of the arguments for why rRNA sequence information might re-write our concepts of classification of prokaryotes. From this and many of the other papers from Woese and Fox and others before 1977 it had been shown that one could use rRNA sequence information to more accurately infer relationships among “prokaryotes” than had been done previously with other types of information. Today this notion that we can use sequence information to infer the evolutionary history of microbes is taken for granted but back in the early 1970s it was not. And in addition, many people probably just did not care too much about the exact details of microbial phylogeny and classification.
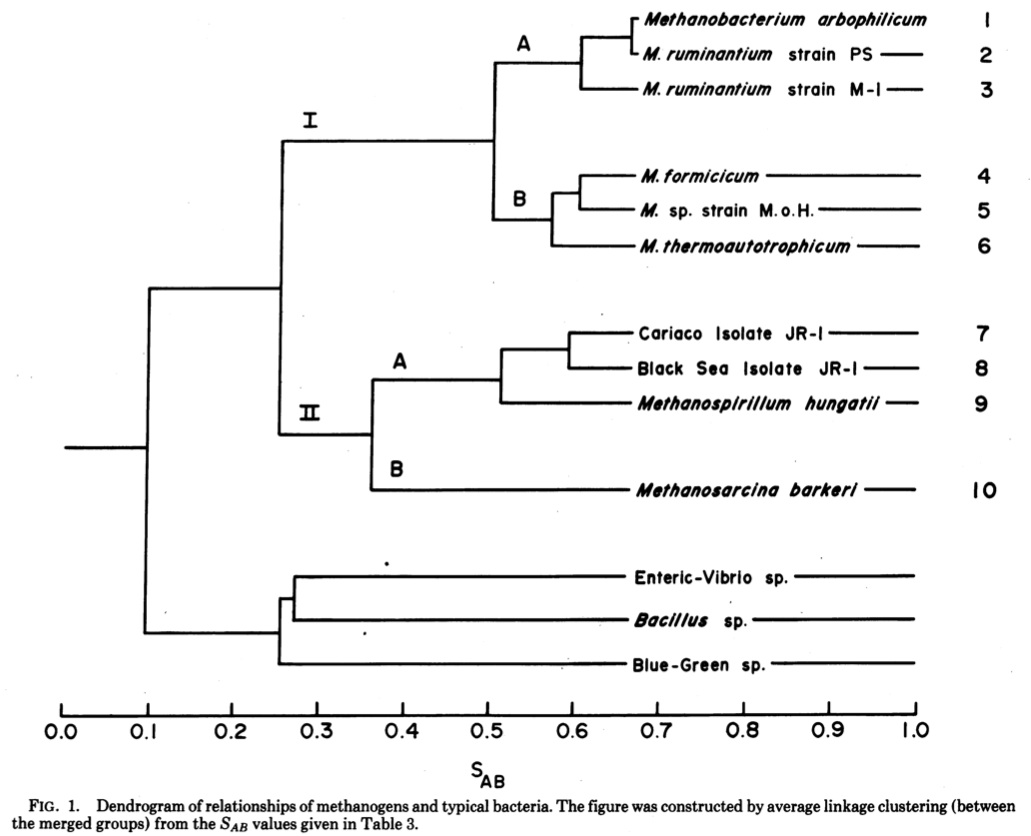
But this changed in the 1977 with that series of papers outlined above. What these papers showed was that hidden beneath everyone’s noses was a separate, previously unknown, major split in the prokaryotes into two distinct lineages. One of these included all the standard bacteria people were familiar with like E. coli and B. subtilis and one of them included some pretty weird wacked out bugs that thrived in extreme conditions. For example, look at the phylogenetic tree from Fox et al.

This tree (made using a distance based clustering algorithm where the distances represent a measure of the similarity of the catalog of short ologonucleotides found in each species) shows the normal bacteria on one side (down below) and methanogens and their relatives on another side. I like the last line of the abstract, which to an evolutionary microbiologist can be considered equivalent to Watson and Crick’s “It has not escaped our notice …”. Here Fox et al. say “These organisms appear to be distantly related to typical bacteria”
The Bach et al. paper has similarly interesting, cool nuggets. However, alas, it is not available in PubMed Central as are the other two papers here I am not focusing on it. What is great though is that the other two papers are freely available to anyone to read in Pubmed Central and also at the PNAS web site. Yay for access. Too bad the other paper is not freely available.
Anyway, fortunately, the most critical of these papers is the Woese and Fox paper from PNAS which is
freely available And it is in this paper that they full argument is laid out. Consider the abstract:
ABSTRACT A phylogenetic analysis based upon ribosomal RNA sequence characterization reveals that living sys.tems represent one of three aboriginal lines of descent: (i) the eubacteria, comprising all typical bacteria; (ii) the archaebacteria, containing methanogenic bacteria; and (iii) the urkaryotes, now represented in the cytoplasmic component of eukaryotic cells.
In this paper they lay out the evidence for the existence of at least three main branches in the Tree of Life. Interestingly, for the phylogenetically minded people out there, they do not show an evolutionary tree in the paper. What they show is what is known as a similarity matrix (the inverse in essence of the distance matrices many people may be used to seeing) where a score is given for the similarity between organisms in the fingerprints of their 16S/18S rRNAs).
If one scans through the matrix one can clearly see three clusters of similarity scores
From this table, Woese and Fox infer the existence of three primary branches in the tree of life. This is laid out in a few paragraphs starting with one at the bottom of page 5088.
A comparative analysis of these data, summarized in Table 1, shows that the organisms clearly cluster into several primary kingdoms. The first of these contains all of the typical bacteria so far characterized …. (lots of names here) … It is appropriate to call this urkingdom the eubacteria.
And then a second paragraph discusses the second group
A second group is defined by the 18S rRNAs of the eukaryotic cytoplasm-animal, plant, fungal, and slime mold (unpublished data). … (They call this lineage the urkaryotes).
And then the third paragraph lays out the revolution:
Eubacteria and urkaryotes correspond approximately to the conventional categories “prokaryote” and “eukaryote” when they are used in a phylogenetic sense. However, they do not constitute a dichotomy; they do not collectively exhaust the class of living systems. There exists a third kingdom which, to date, is represented solely by the methanogenic bacteria, a relatively unknown class of anaerobes that possess a unique metabolism based on the reduction of carbon dioxide to methane (19-21). These “bacteria” appear to be no more related to typical bacteria than they are to eukaryotic cytoplasms. Although the two divisions of this kingdom appear as remote from one another as blue-green algae are from other eubacteria, they nevertheless correspond to the same biochemical phenotype. The apparent antiquity of the methanogenic phenotype plus the fact that it seems well suited to the type of environment presumed to exist on earth 3-4 billion years ago lead us tentatively to name this urkingdom the archaebacteria. Whether or not other biochemically distinct phenotypes exist in this kingdom is clearly an important question upon which may turn our concept of the nature and ancestry of the first prokaryotes.
Mind you, the whole paper is worth reading, but those three paragraphs lay out a revolution in how one thinks about the tree of life. Now admittedly, some of our notions of the tree of life have changed since 1977 and there is much more of a feeling of mixing and merging between branches than was appreciated back then. And some definitely feel that the archaebacteria (or archaea as they are known today) are not per se a third branch in the tree of life but rather than there are four or five major branches and that archaea may not in fact be a “monophyletic grouping”. But whether you think archaea truly represent a third branch in the tree of life or not, this paper fundamentally altered how we think about the tree and about microbes. The work was even written up in the
New York Times and got a lot of press (not that that is proof of anything – but it got microbial phylogeny into the public’s mind).
I think it is worth having all biology students read and understand this paper. Which is why I now try to cover it in basically all classes whenever I can. I could go on and on, but I will simply end with their last paragraph:
With the identification and characterization of the urkingdoms we are for the first time beginning to see the overall phylogenetic structure of the living world. It is not structured in a bipartite way along the lines of the organizationally dissimilar prokaryote and eukaryote. Rather, it is (at least) tripartite, comprising (i) the typical bacteria, (ii) the line of descent manifested in eukaryotic cytoplasms, and (iii) a little explored grouping, represented so far only by methanogenic bacteria.
Citations
Woese CR, & Fox GE (1977). Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proceedings of the National Academy of Sciences of the United States of America, 74 (11), 5088-90 PMID: 270744
Fox GE, Magrum LJ, Balch WE, Wolfe RS, & Woese CR (1977). Classification of methanogenic bacteria by 16S ribosomal RNA characterization. Proceedings of the National Academy of Sciences of the United States of America, 74 (10), 4537-4541 PMID: 16592452
Balch WE, Magrum LJ, Fox GE, Wolfe RS, & Woese CR (1977). An ancient divergence among the bacteria. Journal of molecular evolution, 9 (4), 305-11 PMID: 408502
Some related posts











{kind=link}