I am very pleased to have another in my “Story behind the paper” series of guest posts. This one is from my friend and colleague Josh Weitz from Georgia Tech regarding a recent paper of his in BMC Genomics. As I have said before – if you have published an open access paper on a topic related to this blog and want to do a similar type of guest post let me know …
—————————————-
A guest blog by Joshua Weitz, School of Biology and Physics, Georgia Institute of Technology
Summary This is a short, well sort-of-short, story of the making of our paper: “A neutral theory of genome evolution and the frequency distribution of genes” recently published in BMC Genomics. I like the story-behind-the-paper concept because it helps to shed light on what really happens as papers move from ideas to completion. It’s something we talk about in group meetings but it’s nice to contribute an entry in this type of forum. I am also reminded in writing this blog entry just how long science can take, even when, at least in this case, it was relatively fast.
The pre-history The story behind this paper began when my former PhD student, Andrey Kislyuk (who is now a Software Engineer at DNAnexus) approached me in October 2009 with a paper by Herve Tettelin and colleagues. He had read the paper in a class organized by Nicholas Bergman (now at NBACC). The Tettelin paper is a classic, and deservedly so. It unified discussions of gene variation between genomes of highly similar isolates by estimating the total size of the pan and core genome within multiple sequenced isolates of the pathogen Streptococcus agalactiae.
However, there was one issue that we felt could be improved: how does one extrapolate the number of genes in a population (the pan genome) and the number of genes that are found in all individuals in the population (the core genome) based on sample data alone? Species definitions notwithstanding, Andrey felt that estimates depended on details of the alignment process utilized to define when two genes were grouped together. Hence, he wanted to evaluate the sensitivity of core and pan geonme predictions to changes in alignment rules. However, it became clear that something deeper was at stake. We teamed up with Bart Haegeman, who was on an extended visit in my group from his INRIA group in Montpellier, to evaluate whether it was even possible to quantitatively predict pan and core genome sizes. We concluded that pan and core genome size estimates were far more problematic than had been acknowledged. In fact, we concluded that they depended sensitively on estimating the number of rare genes and rare genomes, respectively. The basic idea can be encapsulated in this figure:

The top panels show gene frequency distributions for two synthetically generated species. Species A has a substantially smaller pan genome and a substantially larger core genome than does Species B. However, when one synthetically generates a sample set of dozens, even hundreds of genomes, then the rare genes and genomes that correspond to differences in pan and core genome size, do not end up changing the sample rarefaction curves (seen at the bottom, where the green and blue symbols overlap). Hence, extrapolation to the community size will not necessarily be able to accurately estimate the size of the pan and core genome, nor even which is larger!
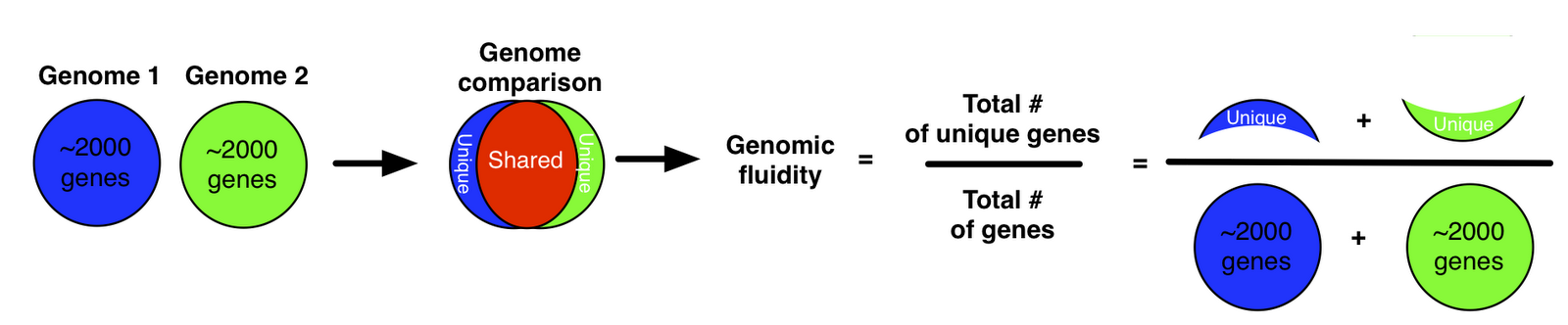
As an alternative, we proposed a metric we termed “genomic fluidity” which captures the dissimilarity of genomes when comparing their gene composition.
The quantitative value of genomic fluidity of the population can be estimated robustly from the sample. Moreover, even if the quantitative value depends on gene alignment parameters, its relative order is robust. All of this work is described in our paper in BMC Genomics from 2011: Genomic fluidity: an integrative view of gene diversity within microbial populations.
However, as we were midway through our genomic fluidity paper, it occurred to us that there was one key element of this story that merited further investigation. We had termed our metric “genomic fluidity” because it provided information on the degree to which genomes were “fluid“, i.e., comprised of different sets of genes. The notion of fluidity also implies a dynamic, i.e., a mechanism by which genes move. Hence, I came up with a very minimal proposal for a model that could explain differences in genomic fluidity. As it turns out, it can explain a lot more.
A null model: getting the basic concepts together
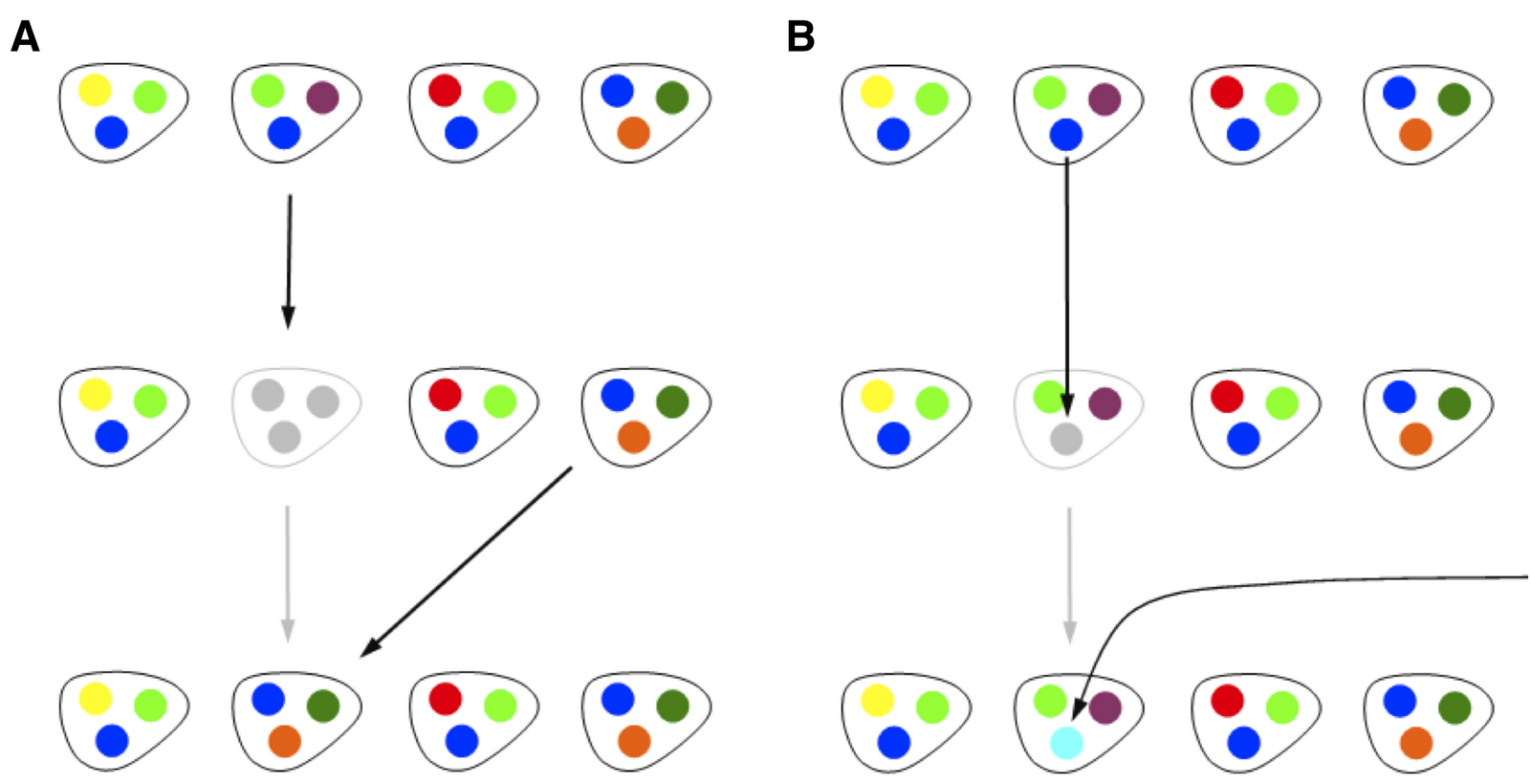
In Spring 2010, I began to explore a minimal, population-genetics style model which incorporated a key feature of genomic assays, that the gene composition of genomes differs substantially, even between taxonomically similar isolates. Hence, I thought it would be worthwhile to analyze a model in which the total number of individuals in the population was fixed at N, and each individual had exactly M genes. Bart and I started analyzing this together. My initial proposal was a very simple model that included three components: reproduction, mutation and gene transfer. In a reproduction step, a random individual would be selected, removed and then replaced with one of the remaining N-1 individuals. Hence, this is exactly analogous to a Moran step in a standard neutral model. At the time, what we termed mutation was actually representative of an uptake event, in which a random genome was selected, one of its genes was removed, and then replaced with a new gene, not found in any other of the genomes. Finally, we considered a gene transfer step in which two genomes would be selected at random, and one gene from a given genome would be copied over to the second genome, removing one of the previous genes. The model, with only birth-death (on left) and mutation (on right), which is what we eventually focused on for this first paper, can be depicted as follows:

We proceeded based on our physics and theoretical ecology backgrounds, by writing down master equations for genomic fluidity as a function of all three events. It is apparent that reproduction decreases genomic fluidity on average, because after a reproduction event, two genomes have exactly the same set of genes. Likewise, gene transfer (in the original formulation) also decreases genomic fluidity on average, but the decrease is smaller by a factor of 1/M, because only one gene is transferred. Finally, mutation increases genomic fluidity on average, because a mutation event occurring at a gene which had before occurred in more than one genome, introduces a new singleton gene in the population, hence increasing dissimilarity. The model was simple, based on physical principles, was analytically tractable, at least for average quantities like genomic fluidity, and moreover it had the right tension. It considered a mechanism for fluidity to increase and two mechanisms for fluidity to decrease. Hence, we thought this might provide a basis for thinking about how relative rates of birth-death, transfer and uptake might be identified from fluidity. As it turns out, many combinations of such parameters lead to the same value of fluidity. This is common in models, and is often referred to as an identifiability problem. However, the model could predict other things, which made it much more interesting.
The making of the paper
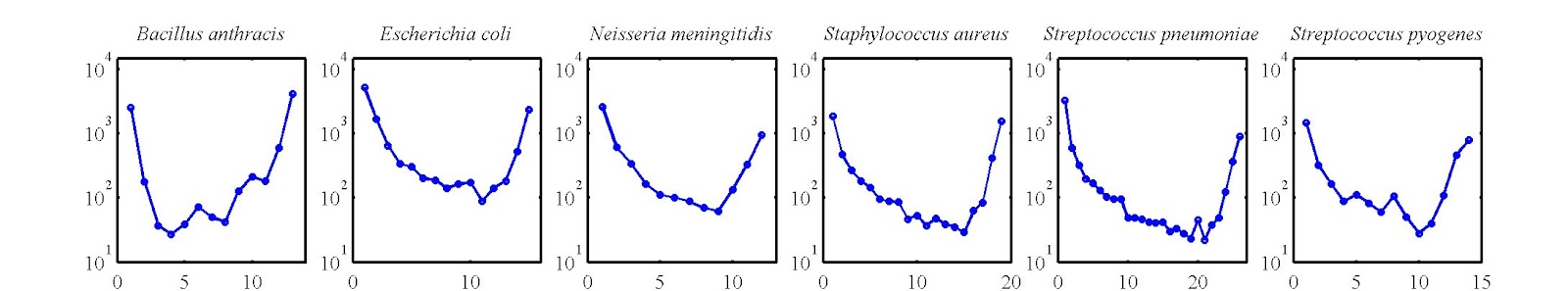
The key moment when the basic model, described above, began to take shape as a paper occurred when we began to think about all the data that we were not including in our initial genomic fluidity analysis. Most prominently, we were not considering the frequency at which genes occurred amongst different genomes. In fact, gene frequency distributions had already attracted attention. A gene frequency distribution summarizes the number of genes that appear in exactly k genomes. The frequency with which a gene appears is generally thought to imply something about its function, e.g., “Comprising the pan-genome are the core complement of genes common to all members of a species and a dispensable or accessory genome that is present in at leastbone but not all members of a species.” (Laing et al., BMC Bioinformatics 2011). The emphasis is mine. But does one need to invoke selection, either implicitly or explicitly, to explain differences in gene frequency?
As it turns out, gene frequency distributions end up having a U-shape, such that many genes appear in 1 or a few genomes, many in all genomes (or nearly all), and relatively few occur at intermediate levels. We had extracted such gene frequency distributions from our limited dataset of ~100 genomes over 6 species. Here is what they look like:

And, when we began to think more about our model, we realized that the tension that led to different values of genomic fluidity also generated the right sort of tension corresponding to U-shaped gene frequency distributions. On the one-hand, mutations (e.g., uptake of new genes from the environment) would contribute to shifting the distribution to the left-hand-side of the U-shape. On the other hand, birth-death would contribute to shifting the distribution to the right-hand side of the U-shape. Gene transfer between genomes would also shift the distribution to the right. Hence, it seemed that for a given set of rates, it might be possible to generate reasonable fits to empirical data that would generate a U-shape. In doing so, that would mean that the U-shape was not nearly as informative as had been thought. In fact, the U-shape could be anticipated from a neutral model in which one need not invoke selection. This is an important point as it came back to haunt us in our first round of review.
So, let me be clear: I do think that genes matter to the fitness of an organism and that if you delete/replace certain genes you will find this can have mild to severe to lethal costs (and occasional benefits). However, our point in developing this model was to try and create a baseline null model, in the spirit of neutral theories of population genetics, that would be able to reproduce as much of the data with as few parameters as possible. Doing so would then help identify what features of gene compositional variation could be used as a means to identify the signatures of adaptation and selection. Perhaps this point does not even need to be stated, but obviously not everyone sees it the same way. In fact, Eugene Koonin has made a similar argument in his nice paper, Are there laws of adaptive evolution: “the null hypothesis is that any observed pattern is first assumed to be the result of non-selective, stochastic processes, and only once this assumption is falsified, should one start to explore adaptive scenarios”. I really like this quote, even if I don’t always follow this rule (perhaps I should). It’s just so tempting to explore adaptive scenarios first, but it doesn’t make it right.
At that point, we began extending the model in a few directions. The major innovation was to formally map our model onto the infinitely many alleles model of population genetics, so that we could formally solve our model using the methods of coalescent theory for both cases of finite population sizes and for exponentially growing population sizes. Bart led the charge on the analytics and here’s an example of the fits from the exponentially growing model (the x-axis is the number of genomes):

At that point, we had a model, solutions, fits to data, and a message. We solicited a number of pre-reviews from colleagues who helped us improve the presentation (thank you for your help!). So, we tried to publish it.
Trying to publish the paper
We tried to publish this paper in two outlets before finding its home in BMC Genomics. First, we submitted the article to PNAS using their new PNAS Plus format. We submitted the paper in June 2011 and were rejected with an invitation to resubmit in July 2011. One reviewer liked the paper, apparently a lot: “I very much like the assumption of neutrality, and I think this provocative idea deserves publication.” The same reviewer gave a number of useful and critical suggestions for improving the manuscript. Another reviewer had a very strong negative reaction to the paper. Here was the central concern: “I feel that the authors’ conclusion that the processes shaping gene content in bacteria and primarily neutral are significantly false, and potentially confusing to readers who do not appreciate the lack of a good fit between predictions and data, and who do not realise that the U-shaped distributions observed would be expected under models where it is selection that determines gene number.” There was no disagreement over the method or the analysis. The disagreement was one of what our message was.
I still am not sure how this confusion arose, because throughout our first submission and our final published version, we were clear that the point of the manuscript was to show that the U-shape of gene frequency distributions provide less information than might have been thought/expected about selection. They are relatively easy to fit with a suite of null models. Again, Koonin’s quote is very apt here, but at some basic level, we had an impasse over a philosophy of the type of science we were doing. Moreover, although it is clear that non-neutral processes are important, I would argue that it is also incorrect to presume that all genes are non-neutral. There’s lots of evidence that many transferred genes have little to no effect on fitness. We revised the paper, including and solving alternative models with fixed and flexible core genomes, again showing that U-shapes are rather generic in this class of models. We argued our point, but the editor sided with the negative review, rejecting our paper in November after resubmission in September, with the same split amongst the reviewers.
Hence, we resubmitted the paper to Genome Biology, which rejected it at the editorial level after a few week delay without much of an explanation, and at that point, we decided to return to BMC Genomics, which we felt had been a good home for our first paper in this area and would likely make a good home for the follow-up. A colleague once said that there should be an r-index, where r is the number of rejections a paper received before ultimate acceptance. He argued that r-indices of 0 were likely not good (something about if you don’t fall, then you’re not trying) and an r-index of 10 was probably not good either. I wonder what’s right or wrong. But I’ll take an r of 2 in this case, especially because I felt that the PNAS review process really helped to make the paper better even if it was ultimately rejected. And, by submitting to Genome Biology, we were able to move quickly to another journal in the same BMC consortia.
Upcoming plans
Bart Haegeman and I continue to work on this problem, from both the theory and bioinformatics side. I find this problem incredibly fulfilling. It turns out that there are many features of the model that we still have not fully investigated. In addition, calculating gene frequency distributions involves a number of algorithmic challenges to scale-up to large datasets. We are building a platform to help, to some extent, but are looking for collaborators who have specific algorithmic interests in these types of problems. We are also in discussions with biologists who want to utilize these types of analysis to solve particular problems, e.g., how can the analysis of gene frequency distributions be made more informative with respect to understanding the role of genes in evolution and the importance of genes to fitness. I realize there are more of such models out there tackling other problems in quantitative population genomics (we cite many of them in our BMC Genomics paper), including some in the same area of understanding the core/pan genome and gene frequency distributions. I look forward to learning from and contributing to these studies.















{kind=link}