Well, this is getting really fun. I have been doing “The Story Behind the Paper” posts for my own papers for a while and recently opened this up to guest posts. And the one today is coming to us from the true wilds – Antarctica. Joe Grzymski (aka @grzymski on Twitter) is out there doing field work (yes, microbiologists have the best field sites …). For more on the field project see the Desert Research Institute’s “Mission Antarctica” site. Joe responded to my request for more guest posts and wrote up a really nice discussion of a recent open access paper of his from the ISME Journal. If anyone else is interesting in writing a guest post on an open access paper or an issue in open access, let me know … without any further ado — below is Joe’s post
Well, this is getting really fun. I have been doing “The Story Behind the Paper” posts for my own papers for a while and recently opened this up to guest posts. And the one today is coming to us from the true wilds – Antarctica. Joe Grzymski (aka @grzymski on Twitter) is out there doing field work (yes, microbiologists have the best field sites …). For more on the field project see the Desert Research Institute’s “Mission Antarctica” site. Joe responded to my request for more guest posts and wrote up a really nice discussion of a recent open access paper of his from the ISME Journal. If anyone else is interesting in writing a guest post on an open access paper or an issue in open access, let me know … without any further ado — below is Joe’s post
I thoroughly enjoy reading Jonathan’s posts detailing – far beyond what can possibly be included in published papers – the who, what, where, when, why and how of science. The story behind the potential fourth domain of life article in PLOS ONE provides great detail about how science is done. After reading Matthew Hahn’s insightful history and commentary on his ortholog conjecture paper I was happy to reply to the request for more “stories” and am chiming in from Antarctica (where I am currently doing field research) to discuss the story behind our recent paper in ISME J, “The significance of nitrogen cost minimization in the proteomes of marine microorganisms”. I hope it will provide another example of how a lot of science is lost in final, streamlined, published versions. Also, it is work that was largely done by an undergraduate and was vigorously and carefully reviewed – the improvements and expansion of ideas because of great reviewers highlights the best of the review process. What started out as a short two-page paper morphed into a larger piece of research – not things you can properly detail in a manuscript.
What was the origin of the idea?
The story behind this paper begins in 1997 when I was in graduate school at Rutgers University. Paul Falkowski joined the faculty right around the time when he published a seminal paper, “The evolution of the nitrogen cycle and its influence on the biological sequestration of CO2 in the ocean.” Paul’s office was across from an office I shared with Jay Cullen (who will factor into the story later); Paul was on my committee and influential in how and what I studied in grad school and as a PostDoc. He constantly kept us on our toes (to say the least). Many of the implications of our recent paper were guided by his thoughts and original work on evolution of the nitrogen cycle and many papers on the functional and ecological factors that dictate the structure of phytoplankton communities. There are many papers here by Paul and the awesome Oscar Schofield- my primary dissertation adviser. Incidentally, I overlapped with Felisa Wolfe-Simon at Rutgers for a few years; she was in the science news recently [#arseniclife], and we had common advisers.
Paul’s paper was pre-genomics – but its scope and breadth are strengthened by recent work on isolates, environmental genomes and transcriptomes from the ocean. Simple mass balance says that the reason why we have oil buried deep in the earth and oxygen in the atmosphere is because photosynthesis (net carbon fixation and oxygenation of the atmosphere) exceeds respiration. During long periods of time, organisms draw down CO2, and it gets sequestered from the atmosphere. In his paper, Paul details an inextricable link between the ratios of nitrogen fixation and denitrification (across geological periods) to the potential draw down of CO2 by particulate organic carbon (namely, large sinking diatoms). That is, if nitrogen fixation is abundant and denitrification is zero, there is more available inorganic nitrogen (in the form of nitrate) in the surface ocean for phytoplankton to utilize and carbon sequestration increases. His paper further details why fixed nitrogen is limiting in the ocean surface across geological scales. It boils down to iron limitation, the specialization required to harness the beastly, triple-bond cracking but woefully inefficient nitrogenase enzyme (which has a high Fe requirement) and also the easier, multiple evolution of the process of denitrification. All of this is articulately summarized here.
How did this work advance?
Fast forward to 2001 and publication of the paper by Baudouin-Cornu et al. In this paper, links between environmental imprinting from fluctuating nutrient availability and atomic composition of assimilatory proteins are quantified. Using genome sequences from E. coli and S. cerevisiae, the authors show that carbon and sulfur assimilatory proteins have amino acid sequences that are depleted in carbon and sulfur side chains, respectively. This makes sense. Proteins high in carbon or nitrogen hardly would provide added fitness to an organism that often struggles to find enough of the nutrient to satisfy other fundamental cellular processes. Similar logic also explains why organisms tend to utilize smaller amino acids more frequently than larger ones: it takes more ATP to make a tyrosine than an alanine. Conversely, the pressure to “cost minimize” is less in organisms, like gut dwelling microbes, that have easy access to amino acids. It is not a perfect rule, but most of the time thermodynamic arguments explain a lot about why organisms do what they do. Fast forward again to Craig Venter’s genomic survey of select surface ocean sites (GOS). This (and now other) sequence data sets provided access to genomic information on organisms that inhabit various surface ocean biomes and, crucially, are largely difficult to isolate in pure culture.
What motivated the writing of the paper?
Last summer, I was sitting in my office writing a proposal. I can’t remember the specific topic, but I was thinking about cost-minimization mostly from the perspective of building proteins in cold environments and the challenges organisms face when it is cold: there is little access to organic carbon (food), and other environmental conditions hamper optimal living. I was re-reading Baudouin-Cornu, and there is a specific sentence in the paper in which the authors hypothesize that the phenomenon of cost-minimization might be a broader evolutionary strategy in resource-limited environments. I figured that organisms that did well in the oligotrophic parts of the ocean probably had mechanisms to reduce nitrogen usage and an easy place to start reducing nitrogen is by not making so many proteins or at the very least reducing the usage of arginine, histadine, lysine, asparagine, tryptophan and glutamine – amino acids with at least one added nitrogen on their side chains.
This is a good spot to introduce my co-author, Alex Dussaq.
 |
| Co-author, Alex Dussaq |
Alex completed his honors undergraduate work in mathematics and biochemistry and was working with me on some coding and analysis projects. To follow Matthew’s example, the conversation that started this paper went like this:
Joe: Alex, I have an interesting idea I want to discuss in a proposal… do you think you can download all the GOS data and calculate the nitrogen, C, H and S atoms per residue side chain as in this paper (hand him Baudouin-Cornu) and then correlate those values with chlorophyll (a proxy for phytoplankton and thus primary productivity), NO3 and Fe. This would be just one figure in the proposal.
Alex: OK, sure that should be pretty easy.
Joe: My proposal is due next week so I need the numbers quickly.
Alex: Yeah, yeah.
Alex codes easier than most people write in their native language. By the way, Alex has moved on to a combined Ph.D./M.D. program at UAB through which he hopes to combine genomics research with new approaches to medicine. I have no doubt he will do unbelievably well in science.
I think that downloading organized data was initially more difficult than it should have been – we spend so much money generating data and so little taking care of it – but we had average values after a few days for several oligotrophic GOS sites and some coastal ocean GOS sites that were convincing enough to put in the proposal. Unfortunately, there are no great metadata – especially physical and chemical characterization of the GOS sites – so we used the “distance to continental land mass” as a proxy for nitrate concentration and oligotrophy (this stung us at first in review). After a week, Alex analyzed all the GOS data and a few important isolated, single organism genomes that factor in the story. After a little less than a month, we had a draft of a two-page brevia that we submitted to Science. It was a simple story that showed data from coastal and open-ocean GOS sites. We found a clear relationship between frequency of nitrogen atoms in side chains of proteins and distance from continental land mass (a proxy for nutrient availability as there are lots of nutrients running off our land). The main conclusion of the paper was that organisms living in oligotrophic oceans tend to have reduced nitrogen content of proteins. Kudos to Alex for some great work.
What was the larger context for the initial findings?
We tried to write the paper from a broader evolutionary and biogeochemical perspective (and used the aforementioned paper by Paul Falkowski as a model). We talked about the implications of organisms in the ocean that are under selective pressure to cost minimize with respect to nitrogen. I’d be happy to share the original submission with anyone who wants to see the evolution of a paper; just contact me. I’d post it here, but Jonathan might charge me for the bytes given how long this is turning out to be. Great reviews make good stories that are decently executed a lot better.
How did the reviewers react?
When reviews of a paper are longer than the original submission, you have an indication that the paper prompted some thought. We received three comprehensive reviews to a two-page paper that contained one main figure and some supplemental material. Given that I didn’t think we could spend time on the subject, we attempted to be brief, too brief especially when compared to the final open access result in ISME. Next, I’ll review some criticisms of the nitrogen cost-minimization hypothesis (having our paper handy will be helpful):
1. Nitrogen cost minimization by simply looking at the predicted proteomes of organisms or environmental genomes assumes that all proteins are made de novo when salvage pathways and dissolved free amino acids (DFAAs) and higher mol. weight/energy compounds are utilized.
Looking at predicted proteomes is indeed a simplification in much the same way that analyzing codon usage frequencies was a simple way to identify with varying degrees of certainty highly expressed genes. No doubt, organisms have multiple methods to acquire the energy they need – especially when under rate-limiting conditions. For example, the pervasive transfer of proteorhodopsin to many different marine microbes presumably helps overcome some nutrient limitation situations by providing added energy from the sun (in the form of a proton gradient), perhaps to aid in transport. The predicted proteome analysis just says that organisms that live in low N waters have lower frequencies of N in their side chains than organisms in the coastal ocean (or in say a sludge metagenome). It doesn’t discount the importance of gene expression, the fact that cells are not “averages” of the genome, etc. None of that really fits into a two-page paper.
2. In our paper, we used the diazotroph Trichodesmium as a model open-ocean organism that was severely N-cost-minimized and compared this to similar success of the SAR11 organism, Pelagibacter ubique. We were criticized because N-fixation should help an organism overcome any N stress.
This was clarified in our next, longer draft. As was shown in the elegant paper by Baudouin-Cornu, assimilatory proteins reflect the “history” of an organism trying to compete for the very atom or molecule they are trying to assimilate. Thus, Trichodesmium would hardly bother to break the triple bond of dinitrogen costing 16 ATP to make ammonia if they were swimming in a vat of inorganic nitrogen. Or put differently, the nitrogenase operon should be nitrogen-cost-minimized reflecting the assimilatory costs of acquiring N. This is, indeed, the case.
3. Why not calculate the bio-energetic costs associated with changes in N content?
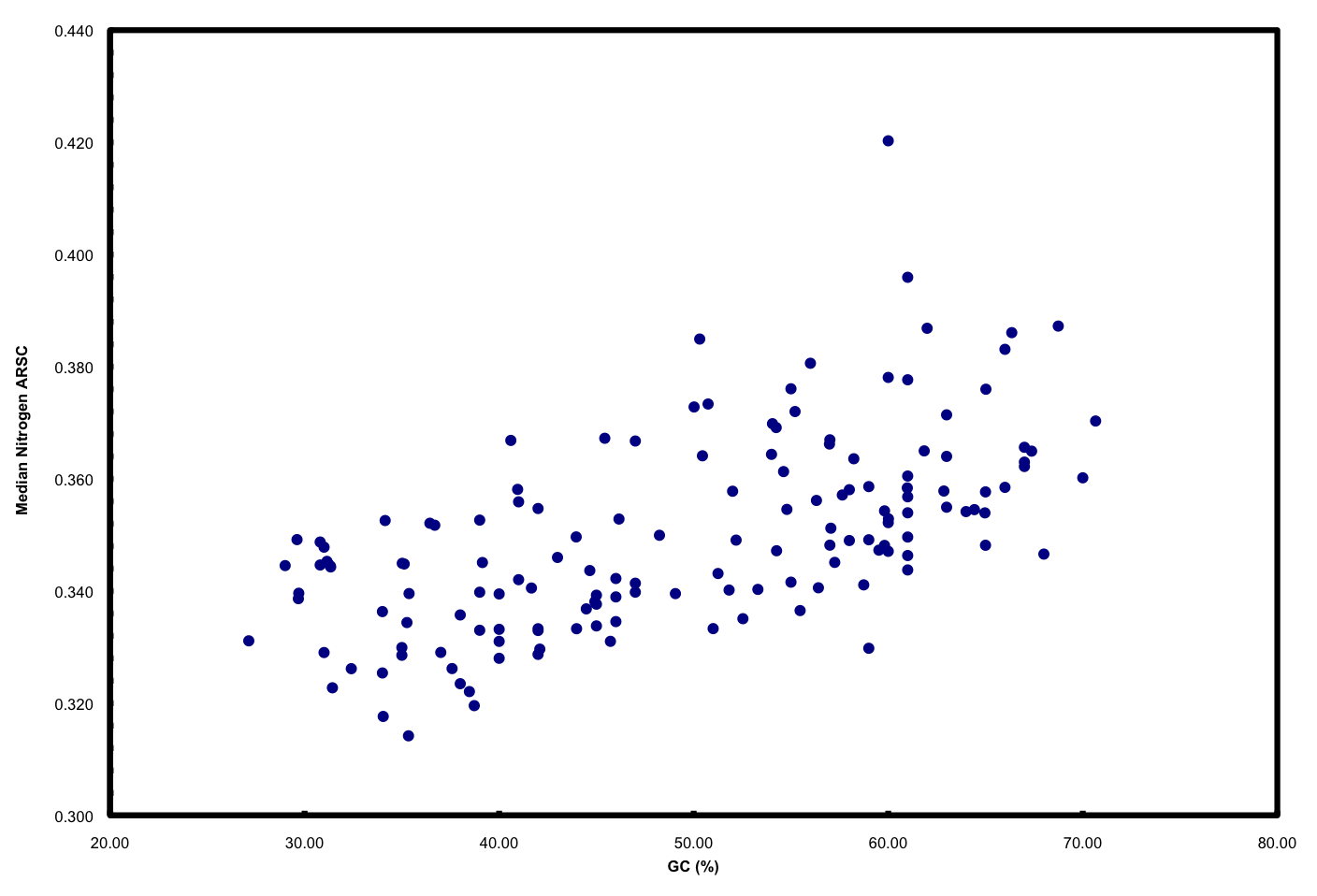
We ended up doing this by proxy in the ISME paper. But it raised a far more interesting point that we pursued in further detail and a chicken/egg argument that was pursued subsequently by another reviewer. If you simply plot N atoms per amino acid side chain versus GC, you get a relationship that looks like this:
This is neither surprising nor novel. But it highlights well the “cost” of having a high GC versus low GC genome in terms of added nitrogen atoms in proteins. These data plotted are all marine microbes but the result is universal.
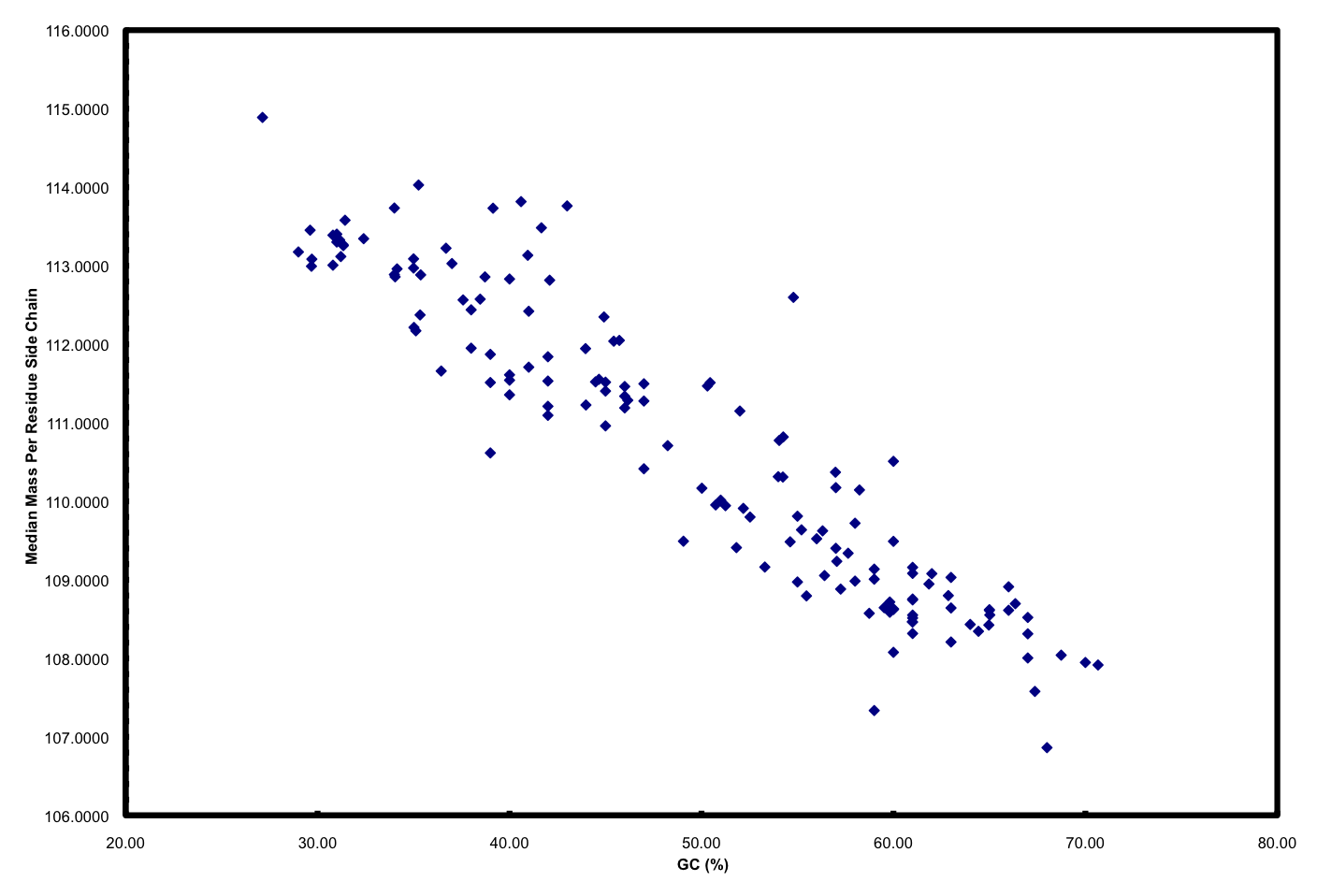
Furthermore, if you plot GC versus median mass of amino acids in the predicted proteome of organisms you get this:
The relationship between GC and the average mass of amino acids is strong. And, this is one of the places where the story gets interesting. Organisms that have low GC genomes have inherently heavier proteins… i.e., All resources being equal and all metabolic pathways being the same (rare, I know), a low GC organism is going to invest more ATP and NADH to make the same protein as a high GC organism. Let’s ignore why this might not matter if you are Helicobacter pylori and quite comfortable acquiring amino acids from your host but focus on ocean microbes. There is a trade-off for all organisms simply based on the GC content of the genome. If you have a low GC genome, you have (on average) larger proteins and less N in your proteins than a high GC genome. Is this trade-off the reason why many of the most successful organisms in the ocean have low GC content? Probably not, but it has to be considered a contributing factor. Constant low nitrogen has to be a major selective pressure given the recent biogeochemical history of the ocean as pointed out in Falkowski (1997). In the final version of the ISME paper, we model differences in the nitrogen budgets of various “model” organisms based on some trade-offs. It was a decent first step, showing that N-cost minimization actually matters.
4. How do you make a quantifiable association between organisms that are so diversely located in space/time and environmental forcing like N availability?
This is a fundamental question in microbial ecology (example, and another). How do we tackle why and when organisms are going to be abundant? Here, I think there are two approaches worth taking. First, what specific genome/metabolic characteristics determine success under specific conditions? For example, what are the characteristics of SAR11 that enable them to “thrive” in oligotrophic waters while their alphaproteobacteria neighbors, the Roseobacter, tend to do better in waters that are more hyper-variable (like the coastal ocean)? Lauro et al. define the characteristics that can be found in genomes of oligotrophic versus copiotrophic organisms. Second, given specific global biogeochemical patterns and environmental forcing constraints, how do we predict organisms will respond? Put in the context of nitrogen cost-minimization, we can ask, “Over geological time will low N waters continue to exert pressure on organisms such that either organisms with N-cost-minimized genomes will thrive or will organisms be forced on a downward GC content trajectory to ease some of this burden?” In our paper, we suggest that the evolutionary history of organisms hints at the impacts nutrient limitations are having on organisms. And this, of course, is by no means new. A beautiful example (albeit not open access).
The divergence of the cyanobacteria Synechococcus and Prochlorococccus during the rise of the diatoms – the most important phytoplankton group in the ocean – suggests the impact of biogeochemical changes on marine microbes. The diversification and proliferation of diatoms in the oceans marginalized cyanobacteria. Diatoms are the workhorses of the ocean biogenic carbon cycle – in comparison to cyanobacteria, they grow quickly and sink faster – thus they sequester fixed CO2, N and Fe that all other surface ocean microbes need. The diatoms changed the ocean, thus putting pressure on cyanobacteria. A result (because many other things also happened) was the genome streamlining and niche adaptation of the lineage. The best example is the high-light adapted MED4 strain of Prochlorococcus. This particular strain has a small genome, low GC and is nitrogen-cost-minimized, as detailed in our paper. Diatoms marginalized cyanobacteria forcing them into specific niches (e.g., high-light, low Fe, low N, low P) where they are successful and well adapted (like these clades that live in iron poor water).
Where we are heading?
What are the implications of cost-minimization in the genomes of ocean microbes? Could it alter the overall nutrient pools in the surface ocean (and thus affect the potential CO2 draw down by phytoplankton)? These are questions we are now pursuing using modeling approaches in an attempt to bolster our understanding of biogeochemistry through genomics and microbial ecology. We are teaming up with Jay Cullen, a chemical oceanography professor, good friend and super smart guy to figure out if cost-minimization and other metabolic changes in microbes might be having more of an effect on biogeochemical cycles than we think. Stay tuned.