
I posted a question to Twitter and Facebook about metrics for assessing density in a phylogenetic tree. Here is a “Storification” of the responses. Thanks for the help all.
Any other suggestions welcome in comments … http://storify.com/phylogenomics/metrics-to-quantify-density-of-taxa-sampling-in-a.js?template=slideshow[View the story “Metrics to quantify density of taxa/sampling in a phylogenetic tree” on Storify]
Tag: Phylogenetics

Phylogenetic analysis of metagenomic data – Mendeley group …
Just a little plug for a Mendeley reference collection I have been helping make on “Phylogenetic and related analyses of metagenomic data.” If you want to know more about such studies you can find a growing list of publications at they group collection. http://www.mendeley.com/groups/1152921/_/widget/29/10/
Phylogenetic and related analyses of metagenomic data is a group in Biological Sciences on Mendeley.
Share this:
Draft post cleanup #21: Tracking progress on the vertebrate tree of life
Yet another post in my “draft blog post cleanup” series. Here is #21; from March 2010:
A very interesting paper came out recently from colleagues of mine at UC Davis: Rapid progress on the vertebrate tree of life. I did not know they were working on this but perhaps should have. It has some fun/interesting analysis of the accumulation of phylogenetic knowledge over time. For example see Figure 1
 |
| Cumulative phylogenetic information amassed for the last 16 years. The accumulation of sequences for vertebrates in GenBank (a), papers using the term ‘phylogeny’ or ‘phylogenetics’ in the Web of Science database (b) and phylogenetic resolution (measured as the proportion of nodes with at least 50% bootstrap support) in the vertebrate tree of life resulting from these research efforts (c). In all cases, the data are cumulative from the start of each analysis. Phylogenetic resolution is calculated as in Table 1. Trend lines are exponential in (a), and second order polynomial in (b) and (c). |
The rest of the paper is worth a look.
And alas I stopped there … I think I wanted to get Brad Shaffer and Bob Thomson’s comments on the paper but never got around to it. Two years later the paper still is worth a look …
Share this:

Draft post cleanup #11: Tree Hugging
Yet another post in my “draft blog post cleanup” series. Here is #11 from September.
Just a quick one. In August a nice review paper came out on phylogenetic analysis software: Learning to Become a Tree Hugger | The Scientist. By Amy Maxmen it is a “A guide to free software for constructing and assessing species relationships”. Definitely worth checking out.
Among the key links & tools discussed:
- Clustal
- RAxML
- MrBayes
- BEAST
- TNT
- BEAST user group
- FigTree
- Mesquite
- Dendroscope
- Paloverde
- GeoPhylo
- MUSCLE
- MAFFT
- T-Coffee
- SeaView
- Phylogeny.fr
- European Bioinformatics Institute
- CIPRES
- Evolutionary Analysis Mesquite
- Biodiverse
- Bayes Traits
- Lagrange
Share this:
I think that I shall never see – metagenomic analysis as lovely as a tree #PhylogenyRules #PLoSOne
![]()
| Figure 2. Phylogenetic tree linking metagenomic sequences from 31 gene families along an oceanic depth gradient at the HOT ALOHA site |
I am a co-author on a new paper that came out in PLoS One yesterday. The paper is PLoS ONE: The Phylogenetic Diversity of Metagenomes and the full citation is Kembel SW, Eisen JA, Pollard KS, Green JL (2011) The Phylogenetic Diversity of Metagenomes. PLoS ONE 6(8): e23214. doi:10.1371/journal.pone.0023214.
The first author is Steven Kembel, a brilliant post doc at the University of Oregon. You can follow him on twitter here. This paper is a product of the “iSEEM” “integrating statistical, ecological and evolutionary approaches to metagenomics” collaboration between my lab and the labs of Jessica Green at U. Oregon and Katie Pollard at UCSF. For more on iSEEM see http://iseem.org. iSEEM was supported by the Gordon and Betty Moore Foundation.
Anyway – the paper focuses on developing and using a new method for assessing the phylogenetic diversity of microbes via in samples via analysis of metagenomic data. Phylogenetic diversity (aka PD) is measured by building evolutionary trees and summing up the total length of branches in such trees. It is an important diversity metric and is complementary to metrics such as “species richness” which is a measure of the number of species in a sample. When one counts species in a sample, one ends up ignoring the evolutionary distances between species and thus one may get an incomplete picture of the diversity of organisms in a sample simply by counting species. For example, a sample that contains 500 different species in the genus Escherichia would have the same “richness” as a sample that contained one representative of each of 500 different Orders of bacteria. For many purposes it is useful to know whether one has a phylogenetically diverse sample or not. (And of course, if one just focuses on species richness it is also important to not simply ignore some set of organisms in the samples as has sort of been done in a recent paper estimating the total species richness on the planet). But that is not the point here – the point here is that counting species, even if done correctly, can give an incomplete picture of the diversity of organisms in sample.
For many years researchers have been attempting to measure phylogenetic diversity of various organisms in various samples. And to do this one needs an evolutionary tree of the organisms in order to then measure branch length in the tree. There is actually a relatively rich history of researchers attempting to look at PD in studies of microbes – especially in cases where one has access to a rRNA tree for the organisms / samples in question. Examples of past work on this include:
- Global patterns in bacterial diversity (by Catherine Lozupone and Rob Knight)
- Status of the microbial census (by Pat Schloss)
- A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea (from my lab and JGI/DSMZ)
- The Cladistic Basis for the Phylogenetic Diversity (PD) Measure …
What we wanted to do here was use metagenomic data to assess phylogenetic diversity of samples. And in particular we wanted to do this with genes other than rRNA genes (e.g., protein coding genes). There were multiple challenges in being able to do this (e.g., see a blog post I made about this issue a few years ago asking for community input). Fortunately, Kembel has worked previously on multiple issues relating to phylogenetic diversity and phylogenetic ecology and his work led to this paper.
I note, as an aside, I have created a Mendeley group focusing on phylogenetic analysis of metagenomes and have added a diversity of papers to the collection:
http://www.mendeley.com/groups/1152921/_/widget/29/2/
Phylogenetic and related analyses of metagenomic data is a group in Biological Sciences on Mendeley.
In the paper Steve basically started with some of the notions and the code from AMPHORA which was designed by Martin Wu (when he was in my lab). AMPHORA automatically infers phylogenetic trees of a set of 31 protein coding genes – and it can do this from genomic or metagenomic data.

AMPHORA was designed to build phylogenetic trees of metagenomic sequences individually – in order to classify reads from samples to infer from what organism they likely came

But that is not what Steven wanted to do here. What he wanted to do was infer phylogenetic trees from metagenomic samples where ALL the organisms in the sample were included in the same tree. This was / is challenging for many reasons and this is what I had written the blog post about previously. One issue we had was the fact that sequences might not overlap with each other and thus including them in a single phylogenetic tree together was complicated.
From my earlier post:
The challenge with this is really two things. First, we want to analyze just the reads themselves (i.e., we do not want to use assemblies you can make from this type of data). Second, and more importantly, we want to include in our analysis sequence reads that only cover small, not necessarily overlapping regions of the “full length” sequence alignments for the family.
The alignment would look something like
- sequence 1 XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
fragment 1 XXXXXXXXX————————-
sequence 2 XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
fragment 2 ———XXXXXXXXXXXX————-
fragment 3 ———————XXXXXXXXXXXXX
fragment 4 —-XXXXXXXXXXXXXXXXXX————
sequence 3 XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
sequence 4 XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
sequence 5 XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
fragment 5 ———————–XXXXXXXXXXX-
- where Xs are the regions covered by the sequences/fragments (could be DNA or amino acids)
We want to build trees from these alignments with the hope of using them to learn lots of cool things about the evolution of the fragments and the species from which they come. I can provide more information but really the key part for the phylogenetics here is the nature of the alignment.
In the past, I have decided to constrain my analyses to NOT deal with this type of alignments. I have either analyzed each fragment on its own or we have built a multiple alignment but only inlcuded fragments that cover more than 3/4 of the full length sequence and thus the matrix is much more filled out. Such an alignment would look like this
- sequence 1 XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
fragment 1 XXXXXXXXXXXXXXXXXXXXXXXXXXX——-
sequence 2 XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
fragment 2 –XXXXXXXXXXXXXXXXXXXXXXXX——–
fragment 3 —–XXXXXXXXXXXXXXXXXXXXXXXXXXXXX
fragment 4 —-XXXXXXXXXXXXXXXXXXXXXXXXXXXX–
sequence 3 XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
sequence 4 XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
sequence 5 XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
fragment 5 –XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX-
But we really want to include the smaller fragments in our analysis. And we are just not certain how to best do this. We know LOTs of people out there think of similar problems in terms of sparse matrices, supermatrices, supertrees, EST data, etc. And we have ideas about how to do this and are asking around by email some phylogenetics gurus we know. But I thought it might be fun to have the discussion on a blog rather than by email.
So again, how might one best build phylogenetic trees from data that looks like this?
- sequence 1 XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
fragment 1 XXXXXXXXX————————-
sequence 2 XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
fragment 2 ———XXXXXXXXXXXX————-
fragment 3 ———————XXXXXXXXXXXXX
fragment 4 —-XXXXXXXXXXXXXXXXXX————
sequence 3 XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
sequence 4 XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
sequence 5 XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
fragment 5 ———————–XXXXXXXXXXX
And from these trees we want to place each fragment relative to (1) the full length sequences and (2) to each other if possible. We also, of course, want branch lengths to reflect some sort of amount of evolution and thus do not just want a cladogram.
So what Steven decided to do in the end was create a method that took all of the AMPHORA markers and concatenated them together into a single mega alignment and then built a reference tree of this mega alignment from available genomes. Then he searched for matches to any of these genes in metagenomic data and built a tree for each sequence that placed it relative to the reference data.
| Figure 1. Conceptual overview of approach to infer phylogenetic relationships among sequences from metagenomic data sets. |
This pipeline allowed him to place many sequences from metagenomic samples onto a single tree such as this one:
| Phylogenetic tree linking metagenomic sequences from 31 gene families along an oceanic depth gradient at the HOT ALOHA site |
And from that he could calculate PD for metagenomic samples. We then used the PD calculations to comparate and contrast PD with other information in particular from the HOT ALOHA metagenomic data set of Ed Delong, Steve Karl and others.
| Figure 3. Taxonomic diversity and standardized phylogenetic diversity versus depth in environmental samples along an oceanic depth gradient at the HOT ALOHA site. |
For more detail on what we did from there on – read the paper. It is open access so all can see it / download it / play with it / whatever. But rather than blather on and on as usual I thought I would email Steve some questions and then post his answers. These are below:
Can you provide any background to how this work got started and why you ended up doing it?
This work got started as a collaboration between the Eisen, Green, and Pollard labs as part of the iSEEM project (“Integrating Statistical Evolutionary & Ecological Approaches to Metagenomics”), which was funded by the Moore Foundation to figure out ways to address ecological and evolutionary questions using metagenomic data. I had a background in using phylogenetic and evolutionary information to understand ecological communities, and one of the things I wanted to do at iSEEM was to try to think about ways that we could apply methods from ecophylogenetics or phylogenetic community ecology to metagenomic data sets. In conversations among the co-authors, we realized that if we could build phylogenetic hypotheses for organisms based on metagenomic data, we could apply a huge body of ecological and evolutionary theory and use these data sets to improve our understanding of microbial communities and their dynamics.
2. How did you end up working on microbes with your background in larger organisms?
The transition from working on macro-organisms to working on microbes actually wasn’t that big of a leap, since my research has generally been question driven rather than study-system or study-organism driven. My previous research involved using phylogenetic information to better understand community assembly in plants and animals. The increasing availability of phylogenetic information for entire communities of plants and animals drove the development of the field of ‘ecophylogenetics’, and it always seeemed to me that microbes would be the ideal system for this type of approach due to the greater availability of sequence data and phylogenetic information for microbes. Also, the development of high-throughput sequencing methods meant that the size of microbial community data sets would quickly become really, really large… the prospect of working on data sets with hundreds of millions of observations was really exciting. As my first postdoc was wrapping up, I collaborated on a study looking at phylogenetic diversity of the rhizobacterial symbionts of plant roots that got me interested in microbial ecology. Right around that time I came across the opportunity to work on the iSEEM project, so it seemed like the perfect opportunity to try a new study system.
Having studied the community ecology of both micro- and macro-organisms, I find it interesting that the fields of microbial and non-microbial phylogenetic community ecology have been fairly insulated from one another until recently. For example, the two fields independently developed phylogenetic approaches to community ecology, each field having its own set of favored statistical methods and software packages, with almost no cross-citation, despite addressing very similar questions. In microbiology the emphasis on phylogenetic diversity measures seems to have been driven by the empirical difficulty of defining microbial ‘species’ and other taxonomic units that macro-organismal ecologists are comfortable with, as well as the availability of phylogenetic and sequence data for microbes. Conversely, for macroorganisms the field of ecophylogenetics was driven by a desire to apply a large body of theory on the links between ecological and evolutionary dynamics to empirical data sets, but was relatively data poor in terms of phylogenetic information about individual species.
3. What was the biggest challenge in this work?
For me the biggest challenge was convincing myself and others that we could infer anything about organismal phylogenies from metagenomic data. People had built phylogenies for individual genes from metagenomic data sets, but there was a lot of skepticism about how and whether it would be possible to infer a phylogeny for multiple genes given the short, non-overlapping nature of metagenomic sequences. A post on your blog provided a lot of useful feedback. In the end this challenge was overcome both through the availability of software packages for placement of short sequences onto reference phylogenies, as well as simulation and bootstrap analyses to make sure that the results we were finding were robust.
4. Any additional things left out of the paper that you would like to mention here? Other acknowledgements? Annoyances?
There were a number of people involved in the iSEEM project, including Samantha Risenfeld and Aaron Darling, who did simulations that were very helpful in figuring out when and whether we could make inferences about phylogenetic relationships among metagenomic reads.
Our paper makes use of a large number of open-source software packages and I’d like to thank the people who made their code available for re-use in this way. In particular the short sequence placement methods implemented in packages like RAxML and pplacer made this study possible.
5. What (in general) are your current and future plans?
Right now I’m working at the Biology & the Built Environment Center on a number of projects studying the phylogenetic and functional diversity of microbes in indoor environments, trying to understand the interaction between architectural design and microbial diversity indoors, and the role indoor microbes play in human health and well being. I am still interseted in plant biology, and I have an ongoing project looking at the diversity and function of microbial communities on plant leaves (the ‘phyllosphere’) in tropical and temperate forests.
Kembel, S., Eisen, J., Pollard, K., & Green, J. (2011). The Phylogenetic Diversity of Metagenomes PLoS ONE, 6 (8) DOI: 10.1371/journal.pone.0023214
Share this:

Phylogeny rules:
I am a coauthor on a new paper in PLoS Computational Biology I thought I would promote here. The full citation for the paper is:
PhylOTU: A High-Throughput Procedure Quantifies Microbial Community Diversity and Resolves Novel Taxa from Metagenomic Data (doi:10.1371/journal.pcbi.1001061).
The paper discusses a new software program “phylOTU” which is for phylogenetic-based identification of “operational taxonomic units”, which are also known as OTUs. What are OTUs? They are basically clusters of closely related sequences that are used to represent something akin to a species. OTUs are used a lot in environmental microbiology b/c one key way to study microbes in the environment is through extraction and sequencing of DNA. Traditionally this has been done through PCR amplification and sequencing of one particular gene (ss-rRNA). Now it is also being done through random sequencing of all DNA from environmental samples (so called metagenomics).
Anyway – the paper is (of course) fully open access and you can read it for more detail. Just thought I would post a little here about it. The paper / project was led by Tom Sharpton, a post doc in Katie Pollard‘s lab at UCSF working on a collaborative project between Katie’s lab, my lab, and Jessica Green‘s lab at U. Oregon (and recently Martin Wu’s new lab at U. Virginia – he was in my lab previously). This collaborative project even has a name “iSEEM” which stands for integrating statistical, evolutionary and ecological approaches to metagenomics. This project has been generously supported by the Gordon and Betty Moore Foundation (via a grant for which I am PI).
Some little tidbits of possible interest about the project
- I really wanted to program to be called POTUS, but I guess I lost out …
- You can get the code here: https://github.com/sharpton/PhylOTU
- You can also get code/data here: http://www.biotorrents.net
Sharpton, T., Riesenfeld, S., Kembel, S., Ladau, J., O’Dwyer, J., Green, J., Eisen, J., & Pollard, K. (2011). PhylOTU: A High-Throughput Procedure Quantifies Microbial Community Diversity and Resolves Novel Taxa from Metagenomic Data PLoS Computational Biology, 7 (1) DOI: 10.1371/journal.pcbi.1001061
Share this:
Summary of #iEVOBIO Day 1 #evolution #phylogenetics #informatics #opensource
Well, just getting around to writing up some thoughts on the iEVOBIO meeting I went to earlier this week. It was really quite excellent so here are some thoughts/notes. Today I am writing about the background and Day 1. Most of this is simply a catalog of what happened along with some twitter details … In a few days I will write up a post on what I think it meant ….
The background: how I heard about iEVOBIO (skip to below if you just want to know about what happened in the meeting)
The first I heard about regarding the meeting was Dec 7, 2009, in a Direct Message on Twitter from @rdmpage. That would be Rod Page, who I had never met, but followed remotely via twitter, his blog, his software and his papers. He wrote
Hi Jonathan, hope you got my email about speaking at iEvoBio in June. No pressure, just checking that it made it into your in box.
I had known about Rod for a long time since I had used his software since I was in grad school. For example, I used to use Treeview for all phylogenetic tree viewing/drawing etc. It seems from the history, it has been available since 1996. Not 100% when I started using it, but it was around then. Then I switched over to using Treeview X a few years later. And I have used on and off some of his other software. More recently I have followed his blog/tweets/web sites closely.
When Rod invited me, I was on a mini vacation in Monterrey and I had not actually seen his email yet (I am ALWAYS behind in reading email). So I found the email, inviting me to give a Keynote at this cool sounding iEVOBIO meeting focusing on informatics for phylogenetics, evolution and biodiversity. Sounded great actually. Especially the part about Open Source:
iEvoBio and its sponsors are dedicated to promoting the practice and philosophy of Open Source software developmentand reuse within the research community. For this reason, if a submitted talk concerns a specific software system for use by the research community, that software must be licensed with arecognized Open Source License, and be available for download, including source code, by a tar/zip file accessed through ftp/http or through a widely used version control system like cvs, Subversion, git, Bazaar, or Mercurial
I also liked the notion of a challenge – in this case there was a challenge for new visualization methods for evolutionary data. In summary the challenge was:
From phylogenetic trees to population networks, whether on printed pages or in GoogleEarth, visualizing evolution is a key part of our discipline. Inspired by the challenges and opportunities visualizing presents for our field, the first iEvoBio challenge is “To create a new visualization tool or platform to support evolutionary science”.
Alas, since I was on vacation I did not have all my schedule information with me, so I said I was not sure. Fortunately, when I got back, it looked like good timing with the Evolution meeting just before so I said sure.
Going to iEVOBIO (skip to below if you just want to know about what happened in the meeting)
Anyway – jump to last week, skipping over some of the preparatory stuff for the meeting. I was planning on being in Oregon for almost a week, including the SSE meeting just before iEVOBIO and a meeting for my iSEEM project in Eugene before that. But I just could not deal with being away for that long including over the weekend, after having not really taken any time off in a while.
So I went home and skipped SSE2010 and then headed back to iEVOBIO on Monday the 28th. I flew on Southwest from Sacramento to Oregon, took the light rail into the city, and walked the last bit to my hotel. I arranged to have dinner with Aaron Darling, a Research Scientists working in/with my lab who was at SSE. We had a good dinner and then I went back to my room and stayed up until about 3:30 AM working on my keynote talk.
I really wanted to include some new stuff and also include some background on microbes and microbial diversity and so worked very late making new slides, piecing together slides from multiple talks, and then trying to delete slides since my talk was way way way too long. The final project I did not finish that night.
I set my alarm on my phone and asked for a wake up call and got about 3 hours sleep. I got up around 6:30 worked on my slides for an hour, and then took a showed and heading downstairs where I borrowed a hotel bike (god, I love Portland – free bikes at my hotel) and biked along the river, over the bridge, and after a little hunting around found where I could park and lock the bike. And I went in.
I worked on my talk for another 30 minutes in an isolated corner and then went over to the main part of the conference center. Finally I was done. I was amazed at how crowded it was. Were all of those people there for iEVOBIO? Alas, no – SSE2010 was still going (I did not realize it would still be on).
After asking around I found the meeting room and met the one and only Rod Page (we had never met). I made sure my laptop would connect with their system and then headed out to get coffee – there was a Starbucks in the hallway outside the meeting room. Alas there was a giant line and my talk was in 25 minutes. Fortunately, Aaron Darling was in the line and he agreed to purchase a latte for me. I went back in, made sure everything was set, and paced around until I got my coffee from Aaron and then it was time for the meeting to start.
The meeting itself: Day1 part 1: keynote by me
The meeting kicked off with a few details from some of the organizers including Rod Page and Todd Vision. We found out who the other organizers were (Rod Page (University of Glasgow), Cecile Ane (University of Wisconsin at Madison), Rob Guralnick (University of Colorado at Boulder), Hilmar Lapp (NESCent), and Cynthia Parr (Encyclopedia of Life). We also found out who helped fund the meetings (US National Evolutionary Synthesis Center (NESCent), and the Society of Systematic Biologists (SSB). I am no longer sure exactly what else they said. But there seems to have been at least one tweet about the intro:
- toranaga Todd Vision mentions how computational biology is a guild, full of people that take great pride in their craft.
And then Rod introduced me. Pretty funny actually. He gave me grief for writing about bad omics words and yet inventing and then using phylogenomics for everything. And then my talk was on. Here it is on slideshare.
Jonathan Eisen talk at #ievobio 2010http://static.slidesharecdn.com/swf/ssplayer2.swf?doc=eisen-100629132410-phpapp01&stripped_title=eisen
View more presentations from Jonathan Eisen.
Also – there were a few tweets about my talk including the following:
- rdmpage IEvoBio starts with keynote by @phylogenomics#ievobio
- toranaga Jonathan Eisen – Phylogenomics of microbes: the dark matter of biology #iEvoBio2010 #iEB10
- cydparr Need @phylogenomics input to Biodiversity Science Triage BoF: using informatics to find the gaps in expertise and knowledge. #ievobio
- toranaga Eisen is recording his own talk. Not out of ego – he’ll put his presentation (and voice recording) on slideshare#iEB10
- toranaga An homage to Donald Rumsfeld by Jonathan Eisen: “There are known knowns. There are known unknowns, there are also unknown unknowns” #iEB10
- toranaga He’s mentions how rRNA has been used to study the diversity of microbes, esp via molecular phylogenetics#iEB10
- toranaga Quote of the conference so far: “Microbes run the planet” – Jonathan Eisen #iEB10
- toranaga @phylogenomics mentions that rRNA analysis doesn’t capture all of the variation in nature, esp at functional level #iEB10
- toranaga @phylogenomics discussing metagenomics and how messy the analysis of it is (this is what I’m trying to solve!) #iEB10
- toranaga A major challenge is the binning of metagenomic data, whereby sequences are sorted into their appropriate genomes #iEB10
- rdmpage Lineage sorting may be major contributor of noise in microbial phylogeny #ievobio
- toranaga Nice talk on microbial diversity from @phylogenomics. Many calls to the community re: assistance building new computational tools. #iEB10
- phylogenomics Here are my slides from my talk at #ievobiohttp://slidesha.re/cZjAso “Phylogenomics of microbes – the dark matter of biology”
- kshameer RT @phylogenomics: Jonathan Eisen talk at#ievobio 2010 http://ff.im/-mUhZi #genomics #evolution#bioinformatics #genetics
- kshameer I can see @nutrigenomics on @phylogenomics#ievobio 2010 slides. Phylogenomics + Nutrigenomics = efficient, nutrition rich probiotics 🙂 ?
- EdwardWinstead Some great slides at the beginning RT @phylogenomics: Jonathan Eisen talk at #ievobio 2010http://bit.ly/9xQKLJ
- nutrigenomics Ha lk 2 C it 2 RT @kshameer: I cn C @nutrigenomics on @phylogenomics #ievobio 2010 slides. Phylogenomics + Nutrigenomics => probiotics+++++
- researchremix Eisen: Analysis of metagenomics is an absolute mess #ievobio
I got asked some great questions afterwards including one by Joe Felsenstein, one by Arlin Stoltzfus, and one by James McInerney (well, McInerney mostly gave me grief about how he disagreed with me about the extent of lateral gene transfer).
Day1 part 2: Short talks
After my talk, thankfully for all involved, there was a coffee break. And then we were back with short, ~15 min talks. These are listed below with some information, most of it from Twitter.
- Vince Smith: Top-down and bottom-up informatics: who has the high ground?
- Vincent Smith discussing scratchpadshttp://scratchpads.eu – looks like neat system for website/research/publication #ievobio
- rdmpage Top down projects have low usage, bottom up have high user base @vsmithuk #ievobio
- toranaga top-down processes tend to be correlated with low usage and slow development. bottom up dependent on user feedback #ievobio
- phylogenomics Smith conclusions: top down projects need institutional support; bottom up depend on many users#ievobio
- phylogenomics #ievobio @vhsvhs says sequential greedy hill climbing is flawed; his simulated annealing method is better
- Cynthia Parr: Community content building for evolutionary biology: Lessons learned from LepTree and Encyclopedia of Life
- toranaga Cynthia Parr – Community content building for evolutionary biology – a talk on LepTree and Encyclopedia of Life #ievobio
- phylogenomics Now @cydparr is comparing LepTree and Encyclopedia of Life #ievobio
- rdmpage #ievobio @cydparr on http://leptree.net
- toranaga http://www.leptree.net/ and http://www.eol.org/for the interested parties. #ievobio
- phylogenomics #ievobio @cydparr showing info from LepTree http://leptree.net including molecular & morphological & fossil data
- phylogenomics #ievobio @cydparr using Drupalhttp://drupal.org/ for LepTree to support community interaction
- toranaga Interesting to see #Drupal playing such an important role in the various biodiversity web tools people are describing at #ievobio
- phylogenomics #ievobio @cydparr built a taxon template for LepTree using semantic tools on top of Drupal
- rdmpage #ievobio @cydparr now talking about @eoflife and LifeDesks
- phylogenomics Semanticizing not useful, communities hard, divide and conqueror scales #ievobio
- #ievobio @cydparr LepTree much more structured DB than EOL
- rdmpage Users have used tools that they asked for @cydparr#ievobio
- rdmpage datadryad @cydparr Reminds us that in biology, the usability adage holds true: Users really don’t know what they want.#ievobio
- cydparr my #ievobio talk is over so now I can really have fun!
- cydparr My #iEvoBio SlideShare upload :Community content building for evolutionary … http://slidesha.re/cbbkmx
- Arlin Stoltzfus: EvoIO: Interop technology meets community science
- phylogenomics NExt up at #ievobio Arlin Stoltzfus from NIST/UMD discussing http://www.evoi.org – informatics/standards for big scale phylogenetics
- rdmpage http://.evoio.org at #ievobio
- toranaga Interoperability is an important consideration when building an infrastructure #ievobio
- phylogenomics #ievobio Stoltzfus discussing Great Fire of Baltimore 1904 & how it lack of interoperability led to standards for fireplugs
- rdmpage http://en.wikipedia.org/wiki/Great_Baltimore_Fireas e.g. Of interop failure #ievobio
- toranaga While standards can be recommended, often times adoption of standard is voluntary. #ievobio
- rdmpage Standards are voluntary, conformance in case of fire hydrants follows disaster #ievobio
- rdmpage Standards developed by stakeholders, compliance is voluntary business decision #ievobio
- phylogenomics #ievobio Standards are voluntary; to further interoperability need to mitigate cost & enhance benefits of compliance; hence EvoIO
- rdmpage http://evoio.org aims to compliance with standards easier #ievobio
- toranaga The EvoIO Stack: Data semantics -> Ontoloties (CDAO), data syntax -> NeXML Format, data access -> phyloWS API #ievobio
- rdmpage http://www.nexml.org/ #ievobio
- rdmpage CDAO http://www.evolutionaryontology.org/#ievobio
- rdmpage Phylowshttps://www.nescent.org/wg/evoinfo/index.php?title=PhyloWS #ievobio
- rdmpage Hackathons at @nascent play a big role in developing these standards #ievobio
- cydparr Arlin Stolzfus is asked “What would evoio.org do with 3 million pounds?” Hackathons, translators, etc. #ievobio
- Brandon Chisham: CDAO-Store: A New Vision for Data Integration
- phylogenomics #ievobio Next up Brandon Chisham @run4ever79 on CDAO Store for Comparative Data Analysis Ontology http://www.evolutionaryontology.org/
- rdmpage CDAO store populated with TreeBase data #ievobio
- rdmpage CDAO-Store queried with Phylows #ievobio
- rdmpage http://www.cs.nmsu.edu/~cdaostore/ #ievobio
- phylogenomics #ievobio CDAO using Prefuse framework for tree viewing searching seehttp://prefuse.org/doc/api/prefuse/data/Tree.html
- rdmpage CDAO future, SPARQL, taxonomy ids, other stores#ievobio
- rdmpage 
- toranaga If anyone was wondering where the CDAO ontology comes from: http://bit.ly/b5TL2w #ievobio
- run4ever79Presentation went well having a working lunch#iEvoBio. Working on phenex integration 🙂
- Joe Felsenstein: Using molecular and morphological data to connect fossils to a phylogeny
- phylogenomics Next up at #ievobio, the one, the only Joe Felsenstein http://bit.ly/bnM4pw on molecular data & paleontology
- rdmpage Joe Felsenstein speaking on fossils and molecular dates #ievobio
- phylogenomics Felsenstein #ievobio discussing how most approaches use synapomorphies/discrete traits and try to date them
- toranaga Joe Felsenstein @ #ievobio on placing fossils on the molecular tree. My old adviser has experience doing this:http://bit.ly/9S8YVS
- phylogenomics Felsenstein presenting a brownian motion model for dating that sounds similar to independent contrasts#ievobio
- cydparr Felsenstein: Brownian motion model for continuous (extant) characters to figure out where to connect fossils to tree. Cool. #ievobio
- toranaga I have a suspicion that Joe Felsenstein dreams in greek #ievobio
- phylogenomics #Now Felsenstein specifically says this dating method is analogous to independent contrasts #ievobio
- rdmpage Felsenstein, simple to compute max likelihood placement of fossil taxa on molecular tree # ievobio
- rdmpage But catch is need calibrated tree #ievobio
- phylogenomics Felsenstein method designed to work if you have a time calibrated molecular clock #ievobio
- rdmpage Now handling case where tree not calibrated#ievobio
- cydparr Felsenstein: Traffic light visualization shows probabilities of potential placements in tree. #ievobio
- rdmpage Missing data in fossils is tiresome #ievobio
- cydparr Felsenstein: Cannot mention non-open-source pub here, cannot thank NSF because they have no clue how to fund methodology #ievobio
- phylogenomics Felsenstein said he was distressed with difficulty in getting funds for software/methods from NSF#ievobio
- toranaga WTF! Funding agencies aren’t picking up Joe Felsenstein’s research?! #ievobio
- phylogenomics Question “How many characters do you need to be precise?” Felsenstein answer “Infinity” #ievobio
- Victor Hanson-Smith: Phylogenetic Mixture Models and Optimization by Simulated Annealing
- toranaga Shoot – was having a conversation about frogs and I missed the title of the next talk. #i‘mabadblogger at#ievobio
- phylogenomics Hanson-Smith making very big statements re possible problems with studies using Max-Likelihood phylogeny b/c of tree search method #ievobio
- rdmpage PhyESTAhttp://markov.uoregon.edu/software/phyesta/ @vhsvhs#ievobio
Day 1: part 3: Lunch – here are some tweets that came out around that time …
- phylogenomics At least evolutionary biologists know what is important: big crowd watching #ESP vs #POR #evol2010#ievobio http://twitpic.com/211kx8
- dgaston83 Lightning talks, Challenge Entries, and Software bazaar coming up at #ievobio
- kcranstn Good idea. Maybe a NeXML / PhyloXML / BEASTXML discussion? RT @rvosa: Plz rt: do we need a NeXML BoF? @rdmpage #ievobio
Day 1 part 4: Challenge talks
And then the post lunch challenge talks began. These related to visualization tools entered into the meeting challenge mentioned above and described here.
- Mike Porter: GenGIS. Here is a link to their submission.
- rdmpage Porter: GenGIS visualizes phylogenies & pie charts on maps, hooks to R for statistics, can record animations. Impressive. #ievobio challenge
- #ievobio challenge up and running. GenGIS http://kiwi.cs.dal.ca/GenGIS/Main_Page and http://dx.doi.org/10.1101/gr.095612.109 up first
- phylogenomics Michael Porter now talking about GenGIS system http://kiwi.cs.dal.ca/GenGIS/Main_Page for mapping biodiversity data #ievobio
- rdmpage GenGIS example of katydids is same @iEvoBio uses for its poster #ievobio
- cydparr rdmpage GenGIS can be scripted using Python #ievobio
- Kris Urie: VoLE (Viewer of Life in EOL). Here is a link to their challenge submission.
- phylogenomics Next up VOLE “Viewer of Life in EOL”#ievobio
- rdmpage Treemap viewer for @eoflife athttp://kurie.github.com/EOL-tree-viewer/ #ievobio
- rdmpage For background entry on @eoflife entry see http://synthesis.eol.org/news/eol-treemap-viewer #ievobio
- cydparr Kris Urie: Viewer of Life on EOL. A treemap, using AJAJ (AJAX + JSON). Shoutout for EOL API. #ievobio challenge
- Arlin Stoltzfus again: Nexplorer3. Here is their challenge entry.
- rdmpage Next challenge entry Nexplorerhttp://exon.niaid.nih.gov/nexplorer/ #ievobio
- rdmpage“Never do live demo in fron of life audience…” @ievobio
- rdmpage Nexplorer3 built upon @cdaotools #ievobio
- cydparr Arlin is back, showing Neplorer3, which uses the EvoIO stack and CDAO, has sparql query window, tree & sequence vis #ievobio challenge
- rdmpageNexplorer3 displays trees, alignments, supports SPARQL #ievobio
- rdmpage Interesting that RDF and SPARQL crop up several times, but nobody has explained what they are #ievobio
- rdmpage Don’t vote for nexplorer3! 😉 Gov’t employee can’t accept money prize #ievobio
- Andrew Hill: PhyloBox. Here is a link to the challenge entry.
- rdmpage Next up Phylobox http://phylobox.appspot.com/viewing trees on the web #ievobio
- phylogenomics Next up Andrew Hill discussing Phylobox – tool for viewing phylogenetic trees in web browserhttp://code.google.com/p/phylobox/ #ievobio
- rdmpage Ah, now I see why phylobox trees were “wonky” #3d#ievobio
- cydparr Andrew Hill: Phylobox, a browser-based phylogeny visualization. With 3D so you can add other data, like video or graphs #ievobio challenge
- phylogenomics Love how in live demo by @andrewxhill tweet by @rdmpage shows up on screen b/c his twitter client was open #ievobio of Phylobox
- rdmpage Phylobox has embeddable widgets #ievobio
- phylogenomics Must say, phyloboxhttp://code.google.com/p/phylobox/ presented by @andrewxhil looks pretty cool #ievobio
- rdmpage How to disrupt a talk using Twitter 😉 #ievobio
- phylogenomics In phylobox, they allow for sharing and lineage tracking for trees; #ievobio
- ryscher
Momma always said: Shut down your twitter client before doing a live demo! #ievobio
- cydparr Hill: Can embed interactive vis widget in a blog or website. Other users can clone and improve — can invite them #ievobio challenge
- rdmpage Phyloxml not #flash so works on #ipad #ievobio
- cydparr Hill: will be merging geophylo and phyloboxhttp://code.google.com/p/phylobox/ should work on iPad#ievobio challenge
- dgaston83 GeoPhylo looks great too from @andrewxhil at#ievobio Google App Engine potentially very powerful for Phylogenetics
- rdmpage OK, now it’s safe to tweet to @andrewxhill #ievobio
- andrewxhill Thanks for keeping it clean for my talk @rdmpage @phylogenomics #ievobio
- rbuels Sitting in #ievobio, was really impressed by Phylobox. So impressed, it got me to tweet.
- phylogenomics Lesson at #ievobio; turn of push notifiers for twitter, email, etc before giving a talk; though seeing tweets about a talk kind of fun
- Sam Smits: jsPhyloSVG. Here is a link to the challenge entry.
- cydparr Smits and Ouverney: library for visualizing vector-based phylogenies on web #ievobio challenge
- rdmpage Next up jsPhyloSVGhttp://phylotouch.jsphylosvg.com/ #ievobio
- phylogenomics Next up at #ievobio Samuel Smiths on Visualizing interactive vector based trees on web; goal to make interactive tool
- rdmpage Fractured web (support for standards, devices) make web development tricky #ievobio
- cydparr I still want to see a user study on effectiveness of circular tree vis. What to do for large trees in print? #ievobio
- toranaga Great point about how diversity of devices and tools frustrates modern web dev #ievobio
- rdmpage Static phylogeny images can’t be mined #ievobio
- rdmpage jsPhyloSVG uses SVG via @RaphaelJS #ievobio
- phylogenomics Smits: created a java script jsPhyloSVG to render trees in SVG #ievobio; scalable; runs on most OS/browsers
- rdmpage jsPhyloSVG written entirely in Javascript #ievobio
- toranaga Using js to circumvent need for a server – lets the browser parse tree file and render image #ievobio
- dgaston83 PhyloTouch, touch enabled for mobile devices. Slick. #ievobio
- dgaston83 PhyloTouch will be available for us Android folks, good to hear’ #ievobio
- phylogenomics Smits: phyloTouchhttp://phylotouch.jsphylosvg.com/ for browsing trees on touch-screen enabled systems #ievobio #cool #verycool
- cydparr Smits: Making tree graphics interactive and searchable , exposing data — all in the markup & javascript interpreted by browser #ievobio
- dgaston83 Some really awesome visualization tools here at#ievobio. Excellent resources for open, collaborative, web-enabled science
Day 1. Part 5. Lightning talks.
- dgaston83 Now time for the lightning talks. Gives me a good idea what to expect for mine tomorrow. Glad I’m on day 2!#ievobio
- toranaga #ievobio lightening talks! 5 minutes and then the gong goes off
- A. Thessen: New Biology: The Data Conservancy and Data Driven Discovery
- phylogenomics Next up at #ievobio Anne Thessen on http://www.dataconservancy.org – working on data sharing methods
- toranaga Anne Thessen asks: “How do we make data sharing part of the normal work flow of the life sciences?” A great, important question. #ievobio
- phylogenomics Thesson types of data: observational, experimental, high throughput, monitoring, simulation#ievobio
- cydparr Lighting talks: Thessen attempts to describe Data Conservancy in five minutes. Good luck! #ievobio
- toranaga I agree with Thessen that data visualization, esp in big data projects, is often key to discovery. #ievobio
- kcranstn arrival of @phylogenomics: #ievobio tweets >>#evol2010 tweets
- toranaga There are not enough power sources here in the#ievobio conference room. Running out of juice!
- rdmpage Curious about what’s happening at #ievobio, check out http://twitter.com/#search?q=ievobio
- B. Gemeinholzer: DNA Bank Network Ð a virtual linkage of natural history collections’ voucher specimens and documentation with physical DNA, sequences, and publications
- rdmpage Gemeinholzer: molecular data metadata frequently missing from studies; linkage to vouchers also limited #ievobio
- phylogenomics Next up: Birgit Gemeinholzerhttp://bit.ly/aqmaPt, linking molecular and specimen data#ievobio
- phylogenomics B. Gemeinholzer: DNA Bank Networkhttp://www.dnabank-network.org/ #ievobio
- toranaga I just realized that the Smithsonian logo, currently on a slide at #ievobio, looks a lot like the @BPGlobalPRprofile pic
- M. Porter: iBarcode-nextgen: tools for next generation biodiversity analysis
- phylogenomics Next up Michael Porter on “iBarcode-nextgen: tools for next generation biodiversity analysis”http://www.ibarcode.org/nextgen #ievobio
- omearabrian #ievobio lightning talks would be even more thrilling with a slowly charging van de graaf generator used as an overtime alarm
- phylogenomics Microbes and metagenomics getting many mentions at #ievobio including GOS; binning; phylotyping; rRNA
- C.T. Hittinger: Leveraging skewed transcript abundance by next-generation sequencing to increase the genomic depth of the tree of life
- phylogenomics Next up: C.T. Hittinger on leveraging skewed transcript abundance by next-gen seq to increase the genomic depth of tree of life #ievobio
- rdmpage @robgural running a tight ship at #ievobio#lightningtalks
- phylogenomics First mention of “phylogenomics” as a method for inferring phylogeny using genomes #ievobio
- toranaga Wow, an empirical data-based lightening talk. Impressively efficient used of time from C.T. Hittinger at#ievobio
- cydparr The PhD comics take on starting up a new conference. Ouch! http://bit.ly/cXVCI9 #ievobio
- Susanna Lewis: Functional Gene Ontology Annotation across Species using PAINT
- phylogenomics Next up SuZanna Lewis: Functional Gene Ontology Annotation across Species using PAINT – annotate gene families #ievobio
- phylogenomics Lewis: PAINT “Phylogenetic Annotation INference Tool” allows one to annotate a single gene family across many species #ievobio
- cydparr Suzanna Lewis: propogating protein properties (GO terms) using PAINT . Finally a good use of power of semantics! #ievobio
- rdmpage For moving presentations across computers @dropbox was a saviour #ievobio
- J. Balhoff: Phenex: Ontological Annotation of Phenotypic Diversity
- phylogenomics Next up: J. Balhoff: Phenex: Ontological Annotation of Phenotypic Diversity seehttps://www.phenoscape.org/wiki/Phenex #ievobio
- toranaga Jim Balhoff: “Phenes – Ontological Annotation of Phenotypic Diversity” #ievobio
- phylogenomics And here is the #PLOSOne paper on Phenexhttp://bit.ly/ac5h2P #ievobio
- rdmpage Phenexhttp://dx.doi.org/10.1371/journal.pone.0010500 #ievobio
- cydparr Balhof: Phenex.–> Powerful though assumes biocurators, not biologists. #ievobio
- P. Midford: The Teleost Taxonomy Ontology
- phylogenomics Next up P. Midford: The Teleost Taxonomy Ontology http://bit.ly/a7I7le includes all species in Eschmeyer’s Catalog of Fishes’ #ievobio
- cydparr Midford: Teleost Taxonomy Ontology <– Number of talks including semantic buzzwords is huge. Time to compile refs to prove value? #ievobio
- rdmpage Linnaean taxonomy easier than phylogeny to deal with as an ontology #ievobio
- T.M. Keesey: Toward a Complete Phyloreferencing Language
- rdmpage  Next up @tmkeesey onhttp://namesonnodes.org/app/ #ievobio
- phylogenomics Next up: Mike Keesey: Toward a Complete Phyloreferencing Language (“sort of a SQL for phylogeny”)#ievobio
- rdmpage Downloading Flash 10.1 … … … #ievobio
- phylogenomics Keesey is @tmkeesey; working onhttp://namesonnodes.org/app/ h/t @rdmpage #ievobio
- rdmpage OK, Flash 10.1 installed,http://namesonnodes.org/app/ looks nice #ievobio
- R. Buels: GMOD for Evolutionary Biology
- phylogenomics Next up R. Buels & D. Clements on GMOD for Evolutionary Biology #ievobio
- rdmpage Last lightning talk is on GMOD http://gmod.org/#ievobio
- toranaga Yay! A GMOD tag-team presentation at #ievobiowww.gmod.org
- phylogenomics GMOD is making use of Chado “Natural diversity module” http://gmod.org/wiki/Chado #ievobio
- rdmpage Chado Natural Diversity Modulehttp://gmod.org/wiki/Chado_Natural_Diversity #ievobio
- ksclarke interesting to see so many folks (at least 2 out of 5 of the projects in the visualization challenge at #ievobio) using #svg
- cdaotools Great day @iEvoBio a lot of great talks and demos#ievobio
- aanaqvi RT @phylogenomics GMOD having an evo hackathonhttp://gmod.org/wiki/GMOD_Evo_Hackathon_Proposal#ievobio
- run4ever79 Very productive day @ievobio, @cdaotools a lot of interesting talks and demos. Some great stuff to take back.#ievobio
Day 1. Part6. Software bazaar and demos
Then there were was the software bazaar and challenge demonstrations, which alas, I skipped most of because of the lack of sleep the night before. It seemed quite packed in there and I was just exhausted. So I went back to my hotel, riding the bike I had borrowed from the hotel back, slowly, along the river.
Here is what I missed:
- Software bazaar
- W. Berendsohn: The EDIT Platform for Cybertaxonomy
- R.J. Challis: Pipefinder – semantic pipelines made easy
- B. Gemeinholzer: DNA Bank Network Ð a virtual linkage of natural history collections’ voucher specimens and documentation with physical DNA, sequences, and publications
- M.J. FavŽ: eFECTIV: Shape analysis using elliptical harmonics
- T.M. Keesey: Names on Nodes: Automating the Application of Taxonomic Names within a Phylogenetic Context
- S. Lewis: Functional Gene Ontology Annotation across Species using PAINT
- S. McKay: GBrowse_syn
- M. Porter: iBarcode-nextgen: tools for next generation biodiversity analysis
- D. Rosauer: Biodiverse, a tool for spatial analysis of biological diversity
- R. Scherle: The Dryad Digital Repository
- C.L. Strope: indel-Seq-Gen version 2.0
- M. Youngblood: mt-tRNA-Draw
- Challenge demonstrations
- M. Porter: GenGIS
- K. Urie: VoLE (Viewer of Life in EOL)
- V. Gopalan: Nexplorer3
- A. Hill: PhyloBox
- S. Smits: jsPhyloSVG
Summary of Day 1.
Here are some tweets summarizing Day 1:
- oatp Open science and data sharing at Evolution 2010 andiEvoBio: Posted by petersuber to oa.notes oa.biology oa.new oa… http://bit.ly/bOocHh
- chrisfreeland @rdmpage @cydparr sounds like #ievobio is going well & interesting. v. sorry I couldn’t work it in!
- trinaeroberts Demos of Biodiverse, GenGIS, various tree visualizers… very cool stuff at #iEvoBio today!
- justsayinnn someone explain to me how the ievobio wants open source yet they charged for registration???
- justsayinnn data visualization stuff was way cool today at #ievobio
At the end of the day, I had dinner with Steven Kembel and Tom Sharpton (@toronaga) who I work with on a Gordon and Betty Moore Foundation funded project we call iSEEM. Dinner and conversation were great. I then went for a walk along the river and went back to my room.
Share this:
Most important paper ever in microbiology? Woese & Fox, 1977, discovery of archaea
Well, today in my “Microbial phylogenomics” class at UC Davis we are discussing what I think might be the most important paper (well, actually, series of papers) in the history of microbiology. The papers are the ones where Carl Woese, George Fox and colleagues outline the evidence for the existence of a “hidden” third major branch in the tree of life – what is now known as the archaea. The evidence for this third branch was first laid out in a series of papers in 1977 including:
- An ancient divergence among the bacteria. Balch WE, Magrum LJ, Fox GE, Wolfe RS, Woese CR.J Mol Evol. 1977 Aug 5;9(4):305-11.
- Classification of methanogenic bacteria by 16S ribosomal RNA characterization. Fox GE, Magrum LJ, Balch WE, Wolfe RS, Woese CR. Proc Natl Acad Sci U S A. 1977 Oct;74(10):4537-4541.
-
Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Woese CR, Fox GE. Proc Natl Acad Sci U S A. 1977 Nov;74(11):5088-90.
Now Woese, Fox and others in Woese’s group had been leading up to these publications in ways for years (I note, there were some pretty incredible people involved in these studies in the years before 1977 too including Mitch Sogin, now at MBL, David Stahl, Chuck Kurland, Norm Pace, etc but that is another story). They had been determining the nucletide sequences of small fragments of rRNAs from different species, especially from different organisms that did not have nuclei – the so-called “prokaryotes”. And they were using these sequences to infer the phylogenetic relationships among these microbes.
Consider for example, the paper by SJ Sogin et al in 1972 “Phylogenetic measurement in procaryotes by primary structural characterization. Sogin SJ, Sogin ML, Woese CR. J Mol Evol. 1971;1(1):173-84. This paper laid out some of the arguments for why rRNA sequence information might re-write our concepts of classification of prokaryotes. From this and many of the other papers from Woese and Fox and others before 1977 it had been shown that one could use rRNA sequence information to more accurately infer relationships among “prokaryotes” than had been done previously with other types of information. Today this notion that we can use sequence information to infer the evolutionary history of microbes is taken for granted but back in the early 1970s it was not. And in addition, many people probably just did not care too much about the exact details of microbial phylogeny and classification.
But this changed in the 1977 with that series of papers outlined above. What these papers showed was that hidden beneath everyone’s noses was a separate, previously unknown, major split in the prokaryotes into two distinct lineages. One of these included all the standard bacteria people were familiar with like E. coli and B. subtilis and one of them included some pretty weird wacked out bugs that thrived in extreme conditions. For example, look at the phylogenetic tree from Fox et al.
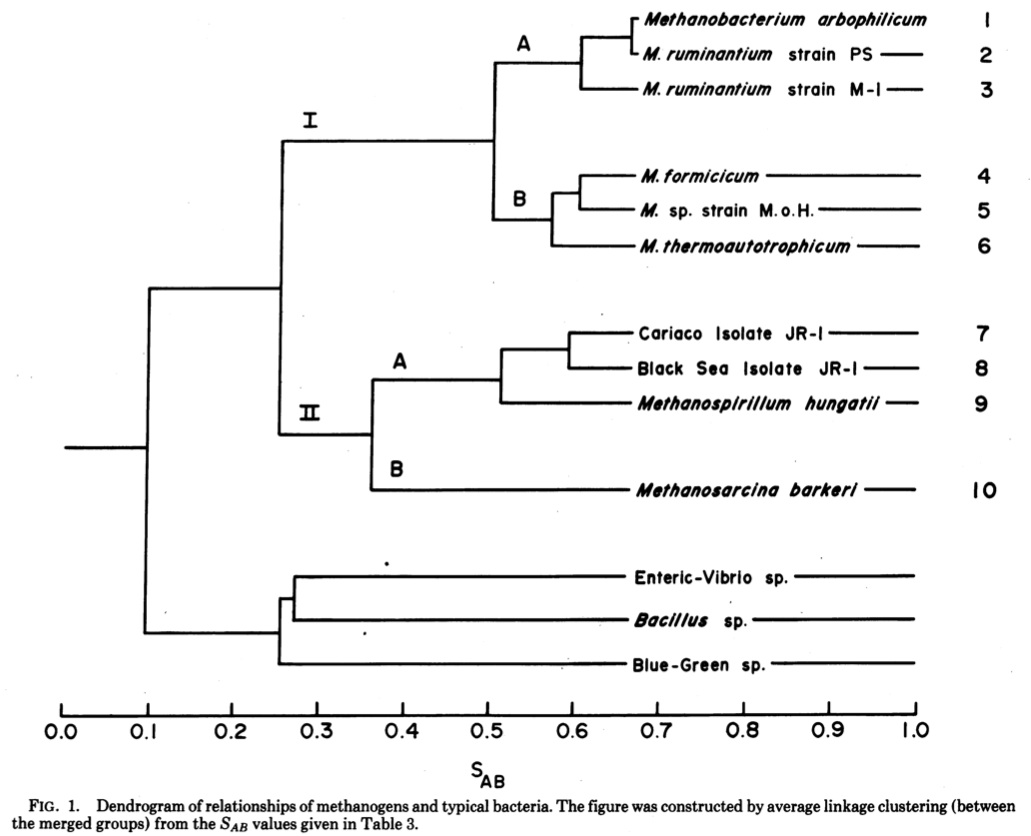
This tree (made using a distance based clustering algorithm where the distances represent a measure of the similarity of the catalog of short ologonucleotides found in each species) shows the normal bacteria on one side (down below) and methanogens and their relatives on another side. I like the last line of the abstract, which to an evolutionary microbiologist can be considered equivalent to Watson and Crick’s “It has not escaped our notice …”. Here Fox et al. say “These organisms appear to be distantly related to typical bacteria”
The Bach et al. paper has similarly interesting, cool nuggets. However, alas, it is not available in PubMed Central as are the other two papers here I am not focusing on it. What is great though is that the other two papers are freely available to anyone to read in Pubmed Central and also at the PNAS web site. Yay for access. Too bad the other paper is not freely available.
Anyway, fortunately, the most critical of these papers is the Woese and Fox paper from PNAS which is freely available And it is in this paper that they full argument is laid out. Consider the abstract:
ABSTRACT A phylogenetic analysis based upon ribosomal RNA sequence characterization reveals that living sys.tems represent one of three aboriginal lines of descent: (i) the eubacteria, comprising all typical bacteria; (ii) the archaebacteria, containing methanogenic bacteria; and (iii) the urkaryotes, now represented in the cytoplasmic component of eukaryotic cells.
In this paper they lay out the evidence for the existence of at least three main branches in the Tree of Life. Interestingly, for the phylogenetically minded people out there, they do not show an evolutionary tree in the paper. What they show is what is known as a similarity matrix (the inverse in essence of the distance matrices many people may be used to seeing) where a score is given for the similarity between organisms in the fingerprints of their 16S/18S rRNAs).

If one scans through the matrix one can clearly see three clusters of similarity scores

From this table, Woese and Fox infer the existence of three primary branches in the tree of life. This is laid out in a few paragraphs starting with one at the bottom of page 5088.
A comparative analysis of these data, summarized in Table 1, shows that the organisms clearly cluster into several primary kingdoms. The first of these contains all of the typical bacteria so far characterized …. (lots of names here) … It is appropriate to call this urkingdom the eubacteria.
And then a second paragraph discusses the second group
A second group is defined by the 18S rRNAs of the eukaryotic cytoplasm-animal, plant, fungal, and slime mold (unpublished data). … (They call this lineage the urkaryotes).
And then the third paragraph lays out the revolution:
Eubacteria and urkaryotes correspond approximately to the conventional categories “prokaryote” and “eukaryote” when they are used in a phylogenetic sense. However, they do not constitute a dichotomy; they do not collectively exhaust the class of living systems. There exists a third kingdom which, to date, is represented solely by the methanogenic bacteria, a relatively unknown class of anaerobes that possess a unique metabolism based on the reduction of carbon dioxide to methane (19-21). These “bacteria” appear to be no more related to typical bacteria than they are to eukaryotic cytoplasms. Although the two divisions of this kingdom appear as remote from one another as blue-green algae are from other eubacteria, they nevertheless correspond to the same biochemical phenotype. The apparent antiquity of the methanogenic phenotype plus the fact that it seems well suited to the type of environment presumed to exist on earth 3-4 billion years ago lead us tentatively to name this urkingdom the archaebacteria. Whether or not other biochemically distinct phenotypes exist in this kingdom is clearly an important question upon which may turn our concept of the nature and ancestry of the first prokaryotes.
Mind you, the whole paper is worth reading, but those three paragraphs lay out a revolution in how one thinks about the tree of life. Now admittedly, some of our notions of the tree of life have changed since 1977 and there is much more of a feeling of mixing and merging between branches than was appreciated back then. And some definitely feel that the archaebacteria (or archaea as they are known today) are not per se a third branch in the tree of life but rather than there are four or five major branches and that archaea may not in fact be a “monophyletic grouping”. But whether you think archaea truly represent a third branch in the tree of life or not, this paper fundamentally altered how we think about the tree and about microbes. The work was even written up in the New York Times and got a lot of press (not that that is proof of anything – but it got microbial phylogeny into the public’s mind).
I think it is worth having all biology students read and understand this paper. Which is why I now try to cover it in basically all classes whenever I can. I could go on and on, but I will simply end with their last paragraph:
With the identification and characterization of the urkingdoms we are for the first time beginning to see the overall phylogenetic structure of the living world. It is not structured in a bipartite way along the lines of the organizationally dissimilar prokaryote and eukaryote. Rather, it is (at least) tripartite, comprising (i) the typical bacteria, (ii) the line of descent manifested in eukaryotic cytoplasms, and (iii) a little explored grouping, represented so far only by methanogenic bacteria.
Citations
Woese CR, & Fox GE (1977). Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proceedings of the National Academy of Sciences of the United States of America, 74 (11), 5088-90 PMID: 270744
Fox GE, Magrum LJ, Balch WE, Wolfe RS, & Woese CR (1977). Classification of methanogenic bacteria by 16S ribosomal RNA characterization. Proceedings of the National Academy of Sciences of the United States of America, 74 (10), 4537-4541 PMID: 16592452
Balch WE, Magrum LJ, Fox GE, Wolfe RS, & Woese CR (1977). An ancient divergence among the bacteria. Journal of molecular evolution, 9 (4), 305-11 PMID: 408502
Some related posts
- The Tree of Life: Some arguments for why Carl Woese (and probably Norm Pace) deserves a Nobel Prize
- Starting my lobbying early this year: Woese/Pace deserve Nobel in …
- The Tree of Life: Here’s hoping molecular classification/systematics …
Share this:

Story Behind the Nature Paper on ‘A phylogeny driven genomic encyclopedia of bacteria & archaea’ #genomics #evolution
![]()
Today is a fun day for me. A paper on which I am the senior author is being published in Nature (yes, the Academic Editor in Chief of PLoS Biology is publishing a paper in Nature, more on that below ..). This paper, entitled, “A phylogeny driven genomic encyclopedia of bacteria and archaea” represents a culmination of years of work by many people from multiple institutions. Today in this blog I am going to do my best to tell the story behind the paper – about the people and the process and a little bit about the science.
First, a brief bit about the science in the paper. In this paper, we (mostly people at the Joint Genome Institute, where I have an Adjunct Appointment — but also people in my lab at UC Davis and at the DSMZ culture collection) did a relatively simple thing – we started with the rRNA tree of life as a guide. Then we identified branches in the bacterial and archaeal portions of this tree where there were no genome sequences available (or in progress) (this was done mostly by Phil Hugenholtz, Dongying Wu and Nikos Kyrpides) Next we searched for representatives of these “unsequenced” branches in the DSMZ culture collection (a collection of bacteria and archaea that can be grown in the lab). And we identified in total some 200 of these. And then the DSMZ (under the direction of Hans-Peter Klenk) grew these organisms and sent the DNA to the Joint Genome Institute. And then JGI turned on their genome sequencing muscle and sequenced the genomes of the organisms in the DNA samples. And finally, we spent a good deal of time then analyzing the data asking a pretty simple question – are there any general benefits that come from this “phylogeny driven” approach to sequencing genomes compared to what one might find with sequencing just any random genome (after all, any genome sequence could have some value)? The paper, describes what we found, which is that there are in fact many benefits that come from sequencing genomes from branches in the tree for which genomes are not available.
More on the details of the science below. But first, I want to note that this paper was truly an amazing team effort, with all sorts of people from the JGI in particular, going above and beyond the call of duty to make sure it happened and worked well. And the Department of Energy has been truly phenomenal in my opinion in supporting this project which in the end is not explicitly about “energy” per se but is really about providing a reference set of genomes that should improve the value of all microbial genome data.
Anyway, now for the story behind the story. And be prepared, because this is a bit long. But I think it is important to place this work in a bigger context both in terms of my background as well as some of the background of other people in the project. If you can’t wait for more on the GEBA project then perhaps you should go to some of these links:
- Videos of talks I have given on the project:
- “Genomic Encyclopedia of Bacteria and Archaea (GEBA)”- Jonathan …
- Recent talk I gave at the Sackler NAS “Microbes and Health” meeting
- Podcast of interview of me for ASM’s Meet the scientist
- Stories about GEBA
- Nature News from 11.17.2009
- Stories about our paper
- Nature News
- GenomeWeb “GEBA Researchers Publish Results from Dozens of Bacterial, Archaeal Genomes”
- Ars Technica article “Presenting a genomic encyclopedia of bacteria (and archaea” by John Timmer
- Iddo Friedberg blogged about it
- The OpenHelix Blog on it
- Leonardo Martins blogs about it here and helps translate a Spanish story about the project
- R&D magazine has a post based on the press releases here
- NY Times story by Carl Zimmer here.
- FriendFeed Discussions here (includes a thread about Nature using a Creative Commons license)
And I will post more links as they come up. Below what I try to provide is some of the story behind the story:
My personal interest in applied uses of phylogenetics stage 1: undergraduate preparation at Harvard
As this paper is primarily about an applied use of phylogenetics (in selecting genomes for sequencing), I thought it would be worth going into some of how I personally became a bit obsessed with applied uses of phylogenetics. For me, my obsession began as an undergraduate at Harvard where I got exposed to the value of phylogeny as a tool from many many angles including but not limited to:
- Freshman year taking a course taught by Stephen Jay Gould where Wayne and David Maddison were Teaching Assistant’s and where they were demoing their new phylogenetics software called MacClade
- Sophomore year taking a conservation biology class with Eric Fajer and Scott Melvin where I was exposed to the concept of “phylogenetic diversity” as a tool in assessing conservation plans
- Junior year working in the lab of Fakhri Bazzaz with people like David Ackerly and Peter Wayne who made use of phylogeny as a key tool in their research projects
- Senior year and the year after graduating where I worked in the lab of Colleen Cavanaugh using rRNA based phylogenetic analysis to characterize uncultured chemosynthetic symbionts. I note it was in Colleen’s lab that I also became obsessed you could say with microbes and why they rock.
My personal interest in applied uses of phylogenetics stage 2: graduate school at Stanford
All of this and more gave me a strong passion for phylogeny as a tool. And so when I went to graduate school at Stanford (originally to work with Ward Watt on butterflies, but then I switched to working in Phil Hanawalt‘s lab on the “Evolution of DNA repair genes, proteins and processes“). And while in that lab I become pretty much obsessed with three things, all related to phylogeny.
First, I was interested in whether the rRNA tree of life, which I had used in my studies in Colleen Cavanaugh’s lab (and in my first paper in J. Bacteriology, which, thanks to ASM, is now in Pubmed Central and free at ASM’s site too), was robust or, as some critics argued, was not that useful. This was a critical question since the best way to study the phylogeny of microbes at the time, and also the best way to study uncultured microbes, was to leverage the ability to clone rRNA genes by PCR and then to build evolutionary trees of those rRNA genes. As part of my graduate work, I did a study where I compared the phylogenetic trees of rRNA to trees of another gene from the same species (I chose, recA). Surprisingly, despite the claims that the rRNA tree was not very useful and that different genes always gave different trees, if you compared the two trees ONLY where there was strong support for a particular branching pattern, the trees of the two genes were in fact VERY VERY similar (a finding that had been suggested previously by others, including Lloyd and Sharp)
Second, I also became obsessed with the fact that most of the experimental studies of DNA repair processes were in a very narrow sampling of the phylogenetic diversity of organisms (e.g., at the time, no studies had been done in archaea, and most studies in bacteria were from only two of the many major groups). So I started experimental studies of repair in halophilic archaea in order to help broaden the diversity of studies. And I began to use PCR to try and clone out repair genes from various other species of diverse bacteria and archaea. Alas, as I was doing this, some institute called TIGR was sequencing the complete genomes of organisms I was trying to clone out single genes from. In fact, one of the first organisms I was working on for PCR studies was Archaeoglobus fulgidus. And when I found out TIGR was sequencing the genome, in a project led by non other than the great microbial evolutionary biologist Hans-Peter Klenk (yes, the same one who helped us in this GEBA project). I decided it was silly to try to clone out individual genes by PCR. And thus I began to learn how to analyze genomes.
It was in the course of learning how to analyze genomes that I came up with another applied use of phylogeny. I realized that one should be able to use phylogenetic studies of genes to help in predicting functions for uncharacterized genes as part of genome annotation efforts. And so I wrote a series of papers showing that this in fact worked (I did this first for the SNF2 family of proteins and then alas coined my own omics word “phylogenomics” to describe this integration of genome analysis and phylogenetics and formalized this phylogenomic approach to functional prediction). I note that what I was arguing for was that protein function could be treated like ANY other character trait and one could use character trait reconstruction methods (which I had learned about while playing with that MacClade program) to infer protein functions for unknown proteins in a protein tree. I note that this notion of predicting protein function from a protein tree is completely analogous to (and one could rightfully say stolen from) how researchers studying uncultured microbes were trying to infer properties of microbes from the position of their rRNA genes in the rRNA tree of life (as I had learned in studies of symbioses).
My personal interest in applied uses of phylogenetics stage 3: my plans for a post doc
So as I was wrapping up graduate school I was seeking a way to go beyond what I was doing and combine studies of DNA repair and evolution and microbiology in another way. And I thought I had found a perfect one in a post doc I accepted with A. John Clark at U. C. Berkeley. John was the person who had discovered recA, the gene I had been using for phylogenetic analysis and for structure function studies. And he was working with none other than Norm Pace and a young hotshot in Norm’s lab, Phil Hugenholtz (as well as a few others including Steve Sandler) in trying to use the recA homolog in archaea as a marker for environmental studies of archaea. It sounded literally perfect. And so I was excited to start this job. That was, until I met Craig Venter.
Grabbing the TIGR by the tail
While I had been playing around with data from TIGR in the latter years of my time in graduate school, I also got involved in teaching a fascinating class with David Botstein, Rick Myers, David Cox and others. (As an aside, this class was part of a new initiative I helped design at Stanford on “Science, Math and Engineering” for non science majors – an initiative that was a pet project of non other than Condie Rice who was Provost at the time). Anyway, Rick Myers was serving as a host for one Craig Venter when he came and gave a talk at Stanford and somehow I managed to finagle my way into being invited to go out to dinner with Craig. And at dinner, I proceeded to tell Craig that I thought some of the evolution stuff he was talking about was bogus and I tried to explain some of my work on phylogeny and phylogenomics. Not sure what Craig thought of the cocky PhD student drawing evolutionary trees on napkins, but it eventually got me a faculty job at TIGR and I worked extensively with Craig so it must have been worth something. And so I and my fiancé Maria-Inés Benito (now wife …) moved to Maryland and spent eight great years there (my wife started in MD as a faculty member at TIGR too, but then she left to go to a company called Informax, may it rest in peace).
Most of my work at TIGR focused on a different side of phylogenomics than represented in the GEBA project. At TIGR I focused on the uses of evolutionary analysis as a component to analyzing genomes – from predicting gene function to finding duplications (e.g., see the V. cholerae genome paper) to identifying genes under unusual patterns of mutation or selection to finding organelle derived genes in nuclear genomes (e.g., see this) to studying the occurrence of lateral gene transfer or the lack of occurrence of it to studying genome rearrangement processes.. And sure, every once in a while I worked on a project where the organism was the first in its major branch to have a genome sequenced (e.g., Chlorobi). And I had noted, along with others that there was a big phylogenetic bias in genome sequencing project (e.g., see my 2000 review paper discussing this here).
But that did not really drive my thinking about what genome to actually sequence until TIGR hired a brilliant microbial systematics expert Naomi Ward as a new faculty member. And it was Naomi who kept emphasizing that someone should go about targeting the “undersequenced” groups in the Tree of Life.
NSF Assembling the Tree of Life grant
And so Naomi and I (w/ Karen Nelson and Frank Robb) put together a grant for the NSF’s “Assembling the Tree of Life” program to do just this – to sequence the first genomes from eight phyla of bacteria for which no genomes were available but for which there were cultured organisms. Amazingly we got the grant. And we did some pretty cool things on that project, including sequencing some interesting genomes, and developing some useful new tools for analyzing genomes (e.g., STAP, AMPHORA, APIS). And I was able to hire some amazing scientists to work in my lab on the project including Dongying Wu (the lead author on the GEBA paper) and Martin Wu (who also worked on the GEBA project and is now a Prof. at U. Virginia) and Jonathan Badger. Alas, we did not publish any earth shattering papers as part of this NSF Tree of Life project on analyzing the genomes of these eight organisms, not because there was not some interesting stuff there but for some other reasons. First, I moved to UC Davis and there was a complicated administrative nightmare in transferring money and getting things up and running at Davis on this project so my lab was not really able to work on it for two years (in retrospect, what a f*ING nightmare dealing with the UC Davis grants system was …).
Then, just as things we ready to get restarted, TIGR kind of imploded and many of the people, including Naomi, my CoPI, left (though I note, my moving to Davis was unrelated to the dissolution of TIGR). But perhaps most importantly, there were some actual technical and scientific problems with our dreams of changing the world of microbiology from our phyla sampling project – the science was not quite there. In particular, having a single genome from each of these phyla was simply not enough to get (and show) the benefits that can come from improved sampling of the tree of life. And thus though we have published some cool papers from this project (e.g., see this PLoS One paper on one of the genomes), we all ended up in one way or another, disappointed with the final results.
Davis and JGI: the return of phylogeny to genomic sampling
When I moved to UC Davis I also was offered (and accepted) an Adjunct Appointment at the Joint Genome Institute (JGI). At both places, I envisioned reinventing myself as someone who worked on studying microbes directly in the environment (e.g., with metagenomics) and symbioses (both of which I had started on at TIGR). And in fact, in a way, I have done this, since I got some medium to big grants to work on these issues. I tried diligently to attend weekly meetings at the JGI but it was difficult since technically I was 100% time at UC Davis and was in essence supposed to be at 0% time at JGI. And when JGI hired Phil Hugenholtz to run their environmental genomics/metagenomics work, I was needed less at JGI since, well, Phil was so good. It was great to go over there and interact with Eddy Rubin, Phil Hugenholtz, and Nikos Kyrpides, among others, but it was unclear what exactly I would do there with Phil running the metagenomics show.
And then, like magic, something came up. I went to one of the monthly senior staff meetings at JGI and while we were listening to someone on the speaker phone, Eddy Rubin handed me a note asking me what I thought about the proposal someone was making to sequence all the species in the Bergey’s Manual. And the light bulb of phylogeny went back on in my head. I told him (I think I wrote it down, but may have said out loud), something like “well, sequencing all 6000 or so species would be great, but it would be better to focus on the most phylogenetically novel ones first.” And in a way, GEBA was born. Eddy organized some meetings at JGI to discuss the Bergey’s proposal and I argued for a more phylogeny driven approach. And this is where having Phil Hugenholtz and Nikos Kyrpides at JGI was like a perfect storm. You see, both had been lamenting the limited phylogenetic coverage of genomes for years, just like I had. Phil had even written a paper about it in 2002 which we used as part of our NSF Tree of Life proposal. And Nikos too had been diligently working for years to make sure novel organisms were sequenced. So though we went to a meeting to discuss the Bergey’s manual idea, we instead proposed an alternative – GEBA.
And for some months, we pitched this notion to various people including at JGI, DOE, and various advisory boards. And the response was basically – “OK – sounds like it COULD be worth doing – why don’t you do a pilot and TEST if it is worth doing” And so, with support from Eddy Rubin and DOE, that is what we did.
One key limitation – getting DNA
So Phil, Nikos and I and a variety of others starting working on the general plan behind GEBA. But there was one key limitation. How were we going to get DNA from all these organisms? One possibility was to seek out diverse people in the community and have them somehow help us. This had some serious problems associated with it, not the least of which was the worry that we might end up sequencing varieties of organisms that people had in their lab but which nobody else had access to (something Naomi Ward and I had written about as a problem a few years before).
And here came the second perfect storm – none other than Hans-Peter Klenk (yes, the same one who had led some of the early genome sequencing efforts when he was at TIGR), was visiting JGI. And he had a relatively new job – at the German Culture Collection DSMZ (In fact, I should note, I had tried to get a job at TIGR even before I met Venter, since they had a position advertised for a “microbial evolutionary biologist” — but that job went to Klenk). Phil Hugenholtz had asked the Head of DSMZ, Erko Stackebrandt, if they might be interested in helping us grow strains and get DNA but we did not yet have a full collaboration with them. And Erko had suggested we contact Hans-Peter. And in his visit to JGI it became apparent that he would do whatever he could to help us build a collaboration with DSMZ. And thus we had a source of DNA. Even more amazingly to me, they did it all for free.
GEBA begins
And thus began the real work in the project. Phil used his expertise with rRNA databases, especially GreenGenes, to pull out phylogenetic trees of different groups. And Nikos used his expertise as the curator of a database on microbial sequencing projects (called GenomesOnline) to help tag which branches in Phil’s tree had sequenced genomes or ones in progress. And then they looked for whether any of the members of the unsequenced branches had representatives in the DSMZ collection. And with some help from Dongying Wu and me, we came up with a list. And with the help of the JGI “Project Management” team including David Bruce and Lynne Goodwin and Eileen Dalin and others at JGI we developed a protocol for collaborating with DSMZ and getting DNA from them.
And I became the chief cheerleader and administrator of the project, in part since Phil and Nikos were so busy with their other things at JGI. And though I was not always on the ball, the project moved forward and we started to get genomes sequenced using the full strength of the JGI as a genome center. The finishing teams at JGI worked diligently on finishing as many of the genomes as possible. And Nikos’ team at JGI made sure that the genomes were annotated. And I helped make sure that they data release policies were broadly open (which everyone at JGI supported). And after many false starts with papers on the project that were way way way to cumbersome and big, with some kicks in the pants from the director of JGI Eddy Rubin who was getting anxious about the project, we turned out the GEBA paper that was published today in Nature.
You might ask, why, as a PLoS official and PLoS cheerleader, we ended up having a paper in Nature? Well, in the end, though I am senior author on the paper, the total contribution to the work mostly came from people at JGI who did not work for me but instead worked with me on this great project. And we took some votes and had some discussions and in the end, despite my lobbying to send it to PLoS Biology, submitting it to Nature was the group decision. I supported this decision in part due to the fact that Nature uses a Creative Commons license for genome papers. But I also supported it because in the end, this was a collaboration involving many many many people and in such projects everyone has to compromise here and there. Now mind you, I am not sad to have a paper in Nature. But I would personally have preferred to have it in a journal that was fully open access, not just occasionally open like Nature.
Now I note, there were a million other things that went on associated with the GEBA project. Some of which I was not even involved in in any way. I will try to write about some of these another time, but this post is already way way way too long. So I am going to just stop here and add that I have been honored and lucky work with people like Phil, Nikos, Hans-Peter, and others on this project and to have the people at the JGI work so hard on the background parts of this project. Thanks to all of them and to the people at DSMZ and in my lab who helped out and to the DOE for funding this work (as well as the Gordon and Betty Moore Foundation, who funded some of the work from my lab on analysis of these genomes). And last but not least, thanks to the Director of JGI Eddy Rubin, supporting this project and for being patient with it and for kicking us in the pants when we needed to get moving on getting a paper out.
Wu, D., Hugenholtz, P., Mavromatis, K., Pukall, R., Dalin, E., Ivanova, N., Kunin, V., Goodwin, L., Wu, M., Tindall, B., Hooper, S., Pati, A., Lykidis, A., Spring, S., Anderson, I., D’haeseleer, P., Zemla, A., Singer, M., Lapidus, A., Nolan, M., Copeland, A., Han, C., Chen, F., Cheng, J., Lucas, S., Kerfeld, C., Lang, E., Gronow, S., Chain, P., Bruce, D., Rubin, E., Kyrpides, N., Klenk, H., & Eisen, J. (2009). A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea Nature, 462 (7276), 1056-1060 DOI: 10.1038/nature08656
Share this:

Story behind the story for new #PLoSOne paper on Bayesian phylogenetics
![]()
There is an interesting new paper in PLoS One” Long-Branch Attraction Bias and Inconsistency in Bayesian Phylogenetics” by Brian Kolaczkowski and Joseph Thornton. The work focuses on methods for inferring phylogenetic history and in particular two types of statistical approaches: Likelihood and Bayesian. These methods are related to each other in that both attempt to use statistical models of evolution and then test different possible phylogenetic trees related taxa by how well certain data sets about those taxa map into the different possible trees. What they did in this new paper was test, with some simulations, and with some mathematical analyses. And somewhat surprisingly, they find that Bayesian methods, which have become more popular recently, appear to be more prone to errors than likelihood methods, when the data sets have multiple not closely related taxa with long branches. (Note if you want to learn more about phylogenetic methods, you can look at the online chapter (html format or PDF) from my Evolution Textbook, though I confess this needs a bit of revision, which I am working on now).
What they see in these cases is that the taxa with long branches group together, something known generally as “Long Branch Attraction” (LBA). Though there have been many previous studies of LBA, most have ended up showing that statistical methods are less prone to this problem than other phylogenetic methods, like distance and parsimony methods. What is surprising in this new work in that they find that Bayesian methods are highly prone to LBA – and much more so than likelihood methods.
Anyway, for more on this one could read the paper. But that I thought might be interesting is to ask the authors for more detail directly. I am hoping to do this more and more with PLoS papers in the future. I was inspired to do this, in fact, by one of the authors of this paper, Joe Thornton. He sent me an email with a link to the paper saying he thought I might be interested in it (true) and that he felt that it was his job in part for a PLoS One paper to make sure it got read by the right audience so he was hoping I might blog about it. And I said sure, but only if he gave me some of the “story behind the story”. So here it is below:
Why did you do these experiments?
Why did we do these experiments? A few years ago, we were studying the behavior of Bayesian posterior probabilities on clades — whether or not they accurately predict the probability that a clade is true, and what kinds of conditions might cause them to deviate from this ideal. We found that when the true tree was in the Felsenstein zone (two non-sister long branches separated by short branches), the long branches were often incorrectly grouped together with strong support. This was just a small part of a much larger paper that was published in MBE in 2008. The suggestion that Bayesian inference (BI) might be biased in favor of a false tree was surprising and intriguing, because we — like most people in the field — had assumed that BI would have the desirable statistical properties of ML (e.g., nearly unbiased inference and statistical consistency — convergence on the true tree with increasing support as the amount of data grows and the evolutionary model is correct, etc.). So we began doing experiments to rigorously explore the nature of the bias and its causes. When we found that BI was statistically inconsistent and the cause was integrating over branch lengths, we knew this result would be controversial, so we wanted to be sure the experiments were truly airtight. We supplemented our initial simulations with analyses of empirical data, with simulations under a wide variety of conditions using all types of priors, as well as mathematical and numerical analyses to clearly demonstrate the reasons for the bias. We also developed software that was identical to fully Bayesian MCMC except that it does not integrate over branch lengths; this method is not subject to the bias that BI displays, clearly demonstrating the cause of the bias.
Why did you send this to PLoS One?
Why did we submit to PLoS One? We think this paper has profound implications for phylogenetic practice and theory, and we want it to have a wide audience. Our experience with the review process in phylogenetic methods, unfortunately, is that many reviewers evaluate manuscripts based on whether or not the results confirm their world-view. This is a legacy of decades of internecine warfare in the field between the adherents of different methodological camps. We write papers in other fields, and while peer-review always has its ups and downs, our experience in phylogenetics is unusual in that solid papers are often rejected for philosophical reasons rather than for reasons of scientific validity and quality. We know this paper will be controversial, and we didn’t want it to be shot down in the review process for partisan reasons. PLoS One seemed like the perfect place to get the paper out and let the scientific community evaluate whether the experiments are convincing or not.
This is our first time publishing in PLoS One. I confess to being a little bit anxious that the paper will be lost in the great tide of papers published in the journal. We know our paper is very strong — I think it’s perhaps the most convincing and complete analysis of any problem I’ve ever published — so we’re confident that the work can have an impact, as long as the attention of readers in the field is drawn to it.
Where is the other author these days?
Bryan is now a postdoc in Andy Kern’s lab at Dartmouth.
Kolaczkowski, B., & Thornton, J. (2009). Long-Branch Attraction Bias and Inconsistency in Bayesian Phylogenetics PLoS ONE, 4 (12) DOI: 10.1371/journal.pone.0007891

