Just an FYI that Holly and I received a grant from Amazon for a bunch of cloud computing credits for the aquarium project. This will allow us to run all of our various 16S analyses for free on their servers.

Just an FYI that Holly and I received a grant from Amazon for a bunch of cloud computing credits for the aquarium project. This will allow us to run all of our various 16S analyses for free on their servers.
Today I did 32 PCR B’s. It may sound like a lot but PCR B’s are extremely easy so it went by pretty quick. I also messed up on updating the google doc and had to throw away three PCR A’s that I did last week. Not a huge deal, but kind of annoying!
Right now we have a lot of Gel Confirmations to do. I counted and there’s 48. So to whoever is in the lab next… have fun! 😉 I’m hoping they all come out good so we can get some sequencing done!
Today I primarily focused on reorganizing some of our stuff. First, I went through our two boxes of extracted DNA and put them in numerical order by sample ID. I think at one point they were in order but with all the PCR redos they’ve gotten quite mixed up and I’ve found it hard and annoying to find the right samples. The first box has any samples under #160 and the second box is any number above #160. The samples are also in order in each box, but I have a feeling that won’t last long 😛
Next I reorganized our PCR box. This didn’t take long. All I did was check to make sure all the forward and reverse primers had stuff in them, replaced a couple empty ones, and put them in order in the box. I did have one concerning discovery though: One of the primers said F2 on the side but F3 on top. I threw that tube away to avoid further confusion.
Lastly, I went through each box of samples from the minus 80 degree freezer and wrote the sample ID range of the samples in that box. For example I would have written “#’s 200-300” for a box with all samples between 200 and 300. It was a bit difficult to write on the boxes because they were so cold!
I’ve spent the week doing PCR A! So far I’m up to 40+ PCR A reactions. I finished up the 20 remaining samples needing PCR A this morning. After losing track of where I was in the PCR A process on Tuesday (and having to restart half of them) I developed an extremely organized way to do today’s set of samples! I meant to take a picture of the chart I made and used and post it here, but I forgot 😦
We are almost done with our preliminary set of samples to be sequenced! Assuming all the PCR A’s work, we will have quite a bit of PCR B to do (which is a lot quicker than PCR A!) and then gel confirmation, quantification, and dilutions to 1 ul. Maybe we can get it done in the next week.. too optimistic? We’ll see!
On a side note, David and a few others get to go to the Kings vs. Spurs game in San Antonio to collect samples for their space project. What a cool opportunity! (Say hi to Tim Duncan for me!)
One thing I would like to do tomorrow is go through our boxes and organize all the DNA, PCR reactions, and final products by sample ID #. I also want to write on the boxes of DNA and PCR reactions which numbers are in that box. For example, we have two DNA boxes (and probably more to come), and it can be quite tedious finding a DNA sample when you don’t know which box it’s in and the ID #’s are written very small on the lids of the 2mL tubes. (You would feel the same way if you had my terrible vision!) I also want to take the original samples out of the freezer and write on the box which samples are in that particular box. Those samples are in the negative 80 degree fridge and it can be quite painful sitting there opening and closing box after box looking for the write samples. Hopefully I get a chance to do that and hopefully it helps 🙂
We will be resuming our weekly lab meetings by hosting Holly’s collaborator, Wesley Grubbs from Pitch interactive. He will be talking about visualization software.
We will be meeting from 1:30 to 3pm in room 4202 of the Genome Center.
Sort of randomly bumped into this one via Google Scholar Alerts I have set up and being from UC Davis the title caught my eye: New World cattle show ancestry from multiple independent domestication events. Yes, we think about cows a lot here OK. And then the second thing that caught my eye were the authors – including David Hillis – who I did not know was working on cattle. I mean – I knew he lived in Texas – so maybe I should have guessed this was coming at some point.
And then I noticed the first author is someone I follow on Twitter. Ok – enough. I got the message. So I looked over the paper.
From the Abstract:
Previous archeological and genetic research has shown that modern cattle breeds are descended from multiple independent domestication events of the wild aurochs (Bos primigenius) ∼10,000 y ago. Two primary areas of domestication in the Middle East/Europe and the Indian subcontinent resulted in taurine and indicine lines of cattle, respectively. American descendants of cattle brought by European explorers to the New World beginning in 1493 generally have been considered to belong to the taurine lineage. Our analyses of 47,506 single nucleotide polymorphisms show that these New World cattle breeds, as well as many related breeds of cattle in southern Europe, actually exhibit ancestry from both the taurine and indicine lineages. In this study, we show that, although European cattle are largely descended from the taurine lineage, gene flow from African cattle (partially of indicine origin) contributed substantial genomic components to both southern European cattle breeds and their New World descendants. New World cattle breeds, such as Texas Longhorns, provide an opportunity to study global population structure and domestication in cattle. Following their introduction into the Americas in the late 1400s, semiferal herds of cattle underwent between 80 and 200 generations of predominantly natural selection, as opposed to the human-mediated artificial selection of Old World breeding programs. Our analyses of global cattle breed population history show that the hybrid ancestry of New World breeds contributed genetic variation that likely facilitated the adaptation of these breeds to a novel environment.
There is some really fascinating stuff in here. And some great figures.
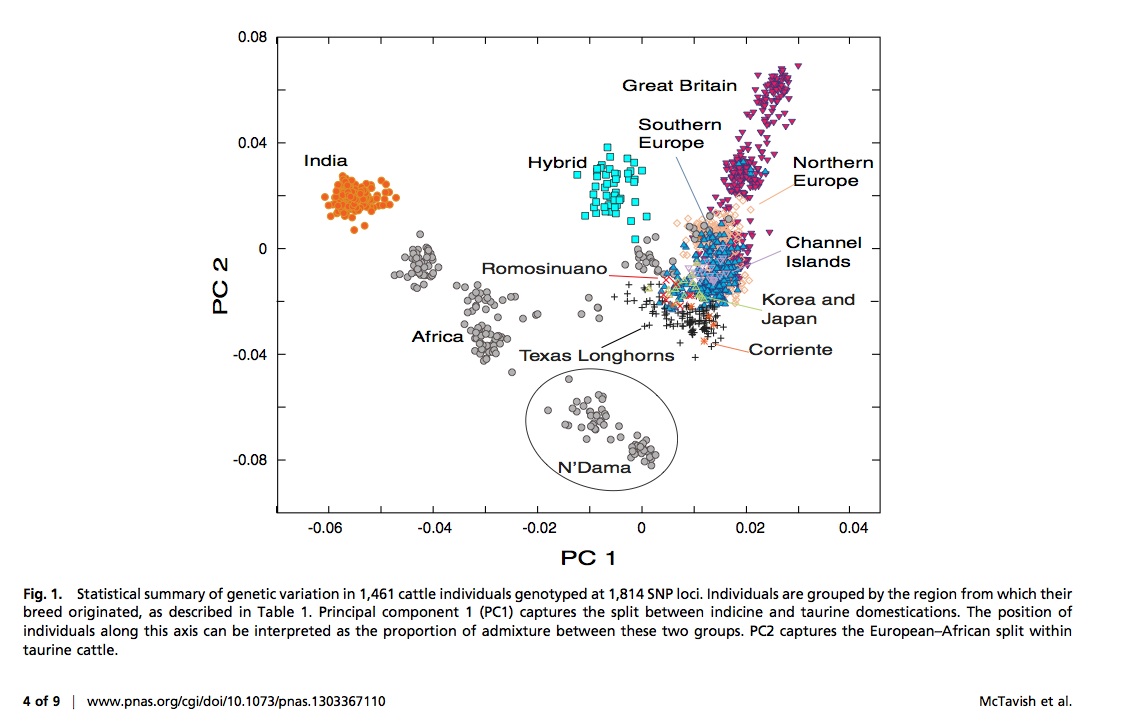
Definitely worth a look if you are interested in cattle, domestication, population genetics, and more …
Interesting paper in Nature Genetics: Genomics of Loa loa, a Wolbachia-free filarial parasite of humans : Nature Genetics : Nature Publishing Group. It is “Open” due to the NPG policy for papers reporting genome data.
Anyway – the paper deals in parts with the biology of the interaction between Wolbachia and filiarial nematodes. Wolbachia are these fascinating intracellular bacteria that are found to infect a diversity of invertebrate species. In 2004 we published the genome sequence of the first Wolbachia genome – a strain that infects Drosophila melanogaster and causes male specific detrimental effects (see summary here and our paper here and a general review here). Many of the Wolbacia that are well studied have male specific effects leading us to jokingly call them “WMDs” the Wolbachia of male destruction.
Interestingly, Wolbachia also infect filarial nematodes, such as the ones that cause various nasty diseases. And these Wolbachia not only do not have any obvious male specific detrimental effects, they appear to be mutualistic symbionts of the nematodes. That is where this paper comes in. The authors sequenced the genome of a filarial nematode that does not have any Wolbachia. The premise here is – if Wolbachia are needed for other nematodes maybe one can figure out what Wolbachia do by identifying features in the Wolbachia-free nematode that are not in the ones with Wolbachia.
They write
Loa loa, the African eyeworm, is a major filarial pathogen of humans. Unlike most filariae, L. loa does not contain the obligate intracellular Wolbachia endosymbiont. We describe the 91.4-Mb genome of L. loa and that of the related filarial parasite Wuchereria bancrofti and predict 14,907 L. loa genes on the basis of microfilarial RNA sequencing. By comparing these genomes to that of another filarial parasite, Brugia malayi, and to those of several other nematodes, we demonstrate synteny among filariae but not with nonparasitic nematodes. The L. loa genome encodes many immunologically relevant genes, as well as protein kinases targeted by drugs currently approved for use in humans. Despite lacking Wolbachia, L. loa shows no new metabolic synthesis or transport capabilities compared to other filariae. These results suggest that the role of Wolbachia in filarial biology is more subtle than previously thought and reveal marked differences between parasitic and nonparasitic nematodes.
Anyway – the paper is worth checking out.
 |
| Figure 3: Phylogenomic analysis of nematodes.. Maximum likelihood, parsimony and Bayesian methods all estimated an identical phylogeny using the concatenated protein sequences of 921 single-copy orthologs. To the left of each node are likelihood bootstrap support values/parsimony bootstrap support values/Bayesian posterior probabilities. The distributions of genes in the ortholog clusters are shown to the right of the phylogeny. Core genes are encoded by all genomes, shared genes are encoded by at least two but fewer than all genomes, and unique genes are found only in one genome. Orthologs specific to the nonparasitic (C. elegans, C. briggsae and P. pacificus) and filarial nematodes are also highlighted. Of the 6,280 L. loa genes with no functional assignment, 3,665 are unique to L. loa and 1,158 are filarial specific. From http://www.nature.com/ng/journal/vaop/ncurrent/full/ng.2585.html |
A couple of recent papers on weight-loss surgery and microbes have gotten a lot of attention to the idea that obesity and microbes have a more than just coincidental connection.
Some of the news stories on the topic are below. A few of them are a bit over the top but the new work seems pretty sound so this is definitely worth a look.
Nice paper from Jonathan Leff and Noah Fierer in PLOS One: Bacterial Communities Associated with the Surfaces of Fresh Fruits and Vegetables
Abstract: Fresh fruits and vegetables can harbor large and diverse populations of bacteria. However, most of the work on produce-associated bacteria has focused on a relatively small number of pathogenic bacteria and, as a result, we know far less about the overall diversity and composition of those bacterial communities found on produce and how the structure of these communities varies across produce types. Moreover, we lack a comprehensive view of the potential effects of differing farming practices on the bacterial communities to which consumers are exposed. We addressed these knowledge gaps by assessing bacterial community structure on conventional and organic analogs of eleven store-bought produce types using a culture-independent approach, 16 S rRNA gene pyrosequencing. Our results demonstrated that the fruits and vegetables harbored diverse bacterial communities, and the communities on each produce type were significantly distinct from one another. However, certain produce types (i.e., sprouts, spinach, lettuce, tomatoes, peppers, and strawberries) tended to share more similar communities as they all had high relative abundances of taxa belonging to the family Enterobacteriaceae when compared to the other produce types (i.e., apples, peaches, grapes, and mushrooms) which were dominated by taxa belonging to the Actinobacteria, Bacteroidetes, Firmicutes, and Proteobacteria phyla. Although potentially driven by factors other than farming practice, we also observed significant differences in community composition between conventional and organic analogs within produce types. These differences were often attributable to distinctions in the relative abundances of Enterobacteriaceae taxa, which were generally less abundant in organically-grown produce. Taken together, our results suggest that humans are exposed to substantially different bacteria depending on the types of fresh produce they consume with differences between conventionally and organically farmed varieties contributing to this variation.
Getting press attention. Examples include:
Definitely worth a look.



