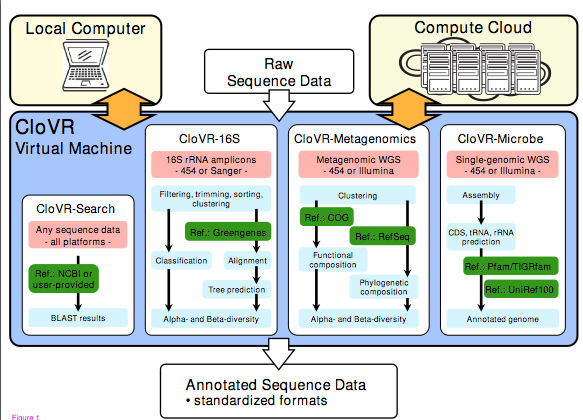
 |
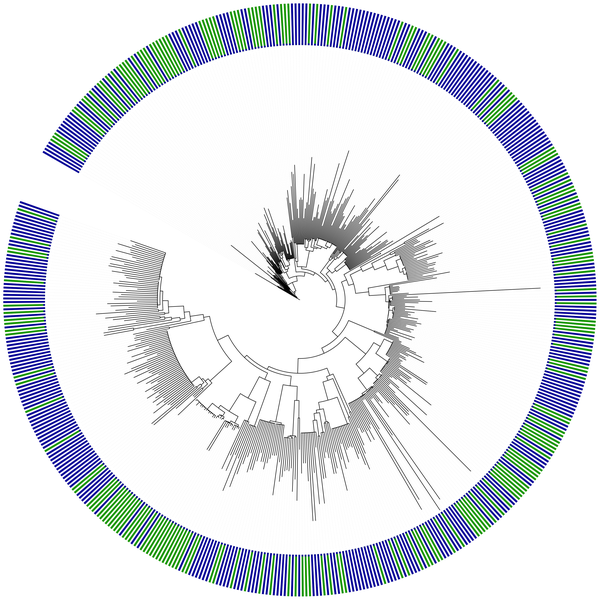
Figure 2. Phylogenetic tree linking
metagenomic sequences from 31 gene
families along an oceanic depth gradient
at the HOT ALOHA site |
I am a co-author on a new paper that came out in PLoS One yesterday. The paper is PLoS ONE: The Phylogenetic Diversity of Metagenomes and the full citation is Kembel SW, Eisen JA, Pollard KS, Green JL (2011) The Phylogenetic Diversity of Metagenomes. PLoS ONE 6(8): e23214. doi:10.1371/journal.pone.0023214.
The first author is Steven Kembel, a brilliant post doc at the University of Oregon. You can follow him on twitter here. This paper is a product of the “iSEEM” “integrating statistical, ecological and evolutionary approaches to metagenomics” collaboration between my lab and the labs of Jessica Green at U. Oregon and Katie Pollard at UCSF. For more on iSEEM see http://iseem.org. iSEEM was supported by the Gordon and Betty Moore Foundation.
Anyway – the paper focuses on developing and using a new method for assessing the phylogenetic diversity of microbes via in samples via analysis of metagenomic data. Phylogenetic diversity (aka PD) is measured by building evolutionary trees and summing up the total length of branches in such trees. It is an important diversity metric and is complementary to metrics such as “species richness” which is a measure of the number of species in a sample. When one counts species in a sample, one ends up ignoring the evolutionary distances between species and thus one may get an incomplete picture of the diversity of organisms in a sample simply by counting species. For example, a sample that contains 500 different species in the genus Escherichia would have the same “richness” as a sample that contained one representative of each of 500 different Orders of bacteria. For many purposes it is useful to know whether one has a phylogenetically diverse sample or not. (And of course, if one just focuses on species richness it is also important to not simply ignore some set of organisms in the samples as has sort of been done in a recent paper estimating the total species richness on the planet). But that is not the point here – the point here is that counting species, even if done correctly, can give an incomplete picture of the diversity of organisms in sample.
For many years researchers have been attempting to measure phylogenetic diversity of various organisms in various samples. And to do this one needs an evolutionary tree of the organisms in order to then measure branch length in the tree. There is actually a relatively rich history of researchers attempting to look at PD in studies of microbes – especially in cases where one has access to a rRNA tree for the organisms / samples in question. Examples of past work on this include:
What we wanted to do here was use metagenomic data to assess phylogenetic diversity of samples. And in particular we wanted to do this with genes other than rRNA genes (e.g., protein coding genes). There were multiple challenges in being able to do this (e.g.,
see a blog post I made about this issue a few years ago asking for community input). Fortunately, Kembel has worked previously on multiple issues relating to phylogenetic diversity and phylogenetic ecology and his work led to this paper.
I note, as an aside, I have created a Mendeley group focusing on phylogenetic analysis of metagenomes and have added a diversity of papers to the collection:
http://www.mendeley.com/groups/1152921/_/widget/29/2/
In the paper Steve basically started with some of the notions and the code from
AMPHORA which was designed by Martin Wu (when he was in my lab). AMPHORA automatically infers phylogenetic trees of a set of 31 protein coding genes – and it can do this from genomic or metagenomic data.
AMPHORA was designed to build phylogenetic trees of metagenomic sequences individually – in order to classify reads from samples to infer from what organism they likely came
But that is not what Steven wanted to do here. What he wanted to do was infer phylogenetic trees from metagenomic samples where ALL the organisms in the sample were included in the same tree. This was / is challenging for many reasons and this is what I had written the blog post about previously. One issue we had was the fact that sequences might not overlap with each other and thus including them in a single phylogenetic tree together was complicated.
From my earlier post:
The challenge with this is really two things. First, we want to analyze just the reads themselves (i.e., we do not want to use assemblies you can make from this type of data). Second, and more importantly, we want to include in our analysis sequence reads that only cover small, not necessarily overlapping regions of the “full length” sequence alignments for the family.
The alignment would look something like
sequence 1 XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
fragment 1 XXXXXXXXX————————-
sequence 2 XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
fragment 2 ———XXXXXXXXXXXX————-
fragment 3 ———————XXXXXXXXXXXXX
fragment 4 —-XXXXXXXXXXXXXXXXXX————
sequence 3 XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
sequence 4 XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
sequence 5 XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
fragment 5 ———————–XXXXXXXXXXX-
where Xs are the regions covered by the sequences/fragments (could be DNA or amino acids)
We want to build trees from these alignments with the hope of using them to learn lots of cool things about the evolution of the fragments and the species from which they come. I can provide more information but really the key part for the phylogenetics here is the nature of the alignment.
In the past, I have decided to constrain my analyses to NOT deal with this type of alignments. I have either analyzed each fragment on its own or we have built a multiple alignment but only inlcuded fragments that cover more than 3/4 of the full length sequence and thus the matrix is much more filled out. Such an alignment would look like this
sequence 1 XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
fragment 1 XXXXXXXXXXXXXXXXXXXXXXXXXXX——-
sequence 2 XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
fragment 2 –XXXXXXXXXXXXXXXXXXXXXXXX——–
fragment 3 —–XXXXXXXXXXXXXXXXXXXXXXXXXXXXX
fragment 4 —-XXXXXXXXXXXXXXXXXXXXXXXXXXXX–
sequence 3 XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
sequence 4 XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
sequence 5 XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
fragment 5 –XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX-
But we really want to include the smaller fragments in our analysis. And we are just not certain how to best do this. We know LOTs of people out there think of similar problems in terms of sparse matrices, supermatrices, supertrees, EST data, etc. And we have ideas about how to do this and are asking around by email some phylogenetics gurus we know. But I thought it might be fun to have the discussion on a blog rather than by email.
So again, how might one best build phylogenetic trees from data that looks like this?
sequence 1 XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
fragment 1 XXXXXXXXX————————-
sequence 2 XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
fragment 2 ———XXXXXXXXXXXX————-
fragment 3 ———————XXXXXXXXXXXXX
fragment 4 —-XXXXXXXXXXXXXXXXXX————
sequence 3 XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
sequence 4 XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
sequence 5 XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX
fragment 5 ———————–XXXXXXXXXXX
And from these trees we want to place each fragment relative to (1) the full length sequences and (2) to each other if possible. We also, of course, want branch lengths to reflect some sort of amount of evolution and thus do not just want a cladogram.
So what Steven decided to do in the end was create a method that took all of the AMPHORA markers and concatenated them together into a single mega alignment and then built a reference tree of this mega alignment from available genomes. Then he searched for matches to any of these genes in metagenomic data and built a tree for each sequence that placed it relative to the reference data.
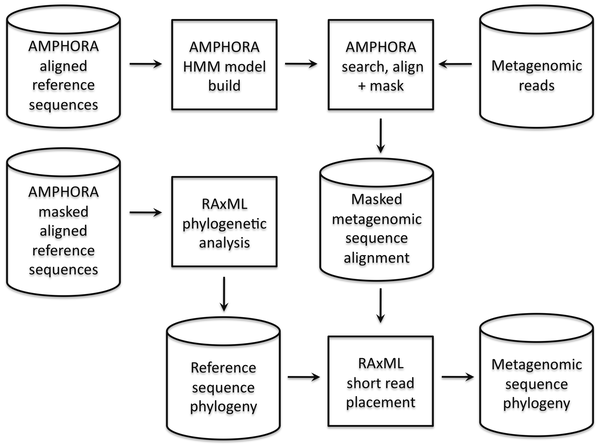 |
| Figure 1. Conceptual overview of approach to infer phylogenetic relationships among sequences from metagenomic data sets. |
This pipeline allowed him to place many sequences from metagenomic samples onto a single tree such as this one:
|
Phylogenetic tree linking metagenomic sequences from 31 gene
families along an oceanic depth gradient at the HOT ALOHA site |
And from that he could calculate PD for metagenomic samples. We then used the PD calculations to comparate and contrast PD with other information in particular from the HOT ALOHA metagenomic data set of Ed Delong, Steve Karl and others.
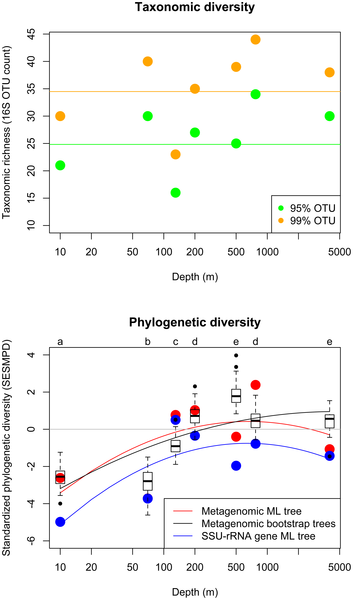 |
Figure 3. Taxonomic diversity and standardized
phylogenetic diversity versus depth in environmental
samples along an oceanic depth gradient at the HOT ALOHA site. |
For more detail on what we did from there on – read the paper. It is open access so all can see it / download it / play with it / whatever. But rather than blather on and on as usual I thought I would email Steve some questions and then post his answers. These are below:
Can you provide any background to how this work got started and why you ended up doing it?
This work got started as a collaboration between the Eisen, Green, and Pollard labs as part of the iSEEM project (“Integrating Statistical Evolutionary & Ecological Approaches to Metagenomics”), which was funded by the Moore Foundation to figure out ways to address ecological and evolutionary questions using metagenomic data. I had a background in using phylogenetic and evolutionary information to understand ecological communities, and one of the things I wanted to do at iSEEM was to try to think about ways that we could apply methods from ecophylogenetics or phylogenetic community ecology to metagenomic data sets. In conversations among the co-authors, we realized that if we could build phylogenetic hypotheses for organisms based on metagenomic data, we could apply a huge body of ecological and evolutionary theory and use these data sets to improve our understanding of microbial communities and their dynamics.
2. How did you end up working on microbes with your background in larger organisms?
The transition from working on macro-organisms to working on microbes actually wasn’t that big of a leap, since my research has generally been question driven rather than study-system or study-organism driven. My previous research involved using phylogenetic information to better understand community assembly in plants and animals. The increasing availability of phylogenetic information for entire communities of plants and animals drove the development of the field of ‘ecophylogenetics’, and it always seeemed to me that microbes would be the ideal system for this type of approach due to the greater availability of sequence data and phylogenetic information for microbes. Also, the development of high-throughput sequencing methods meant that the size of microbial community data sets would quickly become really, really large… the prospect of working on data sets with hundreds of millions of observations was really exciting. As my first postdoc was wrapping up, I collaborated on a study looking at phylogenetic diversity of the rhizobacterial symbionts of plant roots that got me interested in microbial ecology. Right around that time I came across the opportunity to work on the iSEEM project, so it seemed like the perfect opportunity to try a new study system.
Having studied the community ecology of both micro- and macro-organisms, I find it interesting that the fields of microbial and non-microbial phylogenetic community ecology have been fairly insulated from one another until recently. For example, the two fields independently developed phylogenetic approaches to community ecology, each field having its own set of favored statistical methods and software packages, with almost no cross-citation, despite addressing very similar questions. In microbiology the emphasis on phylogenetic diversity measures seems to have been driven by the empirical difficulty of defining microbial ‘species’ and other taxonomic units that macro-organismal ecologists are comfortable with, as well as the availability of phylogenetic and sequence data for microbes. Conversely, for macroorganisms the field of ecophylogenetics was driven by a desire to apply a large body of theory on the links between ecological and evolutionary dynamics to empirical data sets, but was relatively data poor in terms of phylogenetic information about individual species.
3. What was the biggest challenge in this work?
For me the biggest challenge was convincing myself and others that we could infer anything about organismal phylogenies from metagenomic data. People had built phylogenies for individual genes from metagenomic data sets, but there was a lot of skepticism about how and whether it would be possible to infer a phylogeny for multiple genes given the short, non-overlapping nature of metagenomic sequences. A post on your blog provided a lot of useful feedback. In the end this challenge was overcome both through the availability of software packages for placement of short sequences onto reference phylogenies, as well as simulation and bootstrap analyses to make sure that the results we were finding were robust.
4. Any additional things left out of the paper that you would like to mention here? Other acknowledgements? Annoyances?
There were a number of people involved in the iSEEM project, including Samantha Risenfeld and Aaron Darling, who did simulations that were very helpful in figuring out when and whether we could make inferences about phylogenetic relationships among metagenomic reads.
Our paper makes use of a large number of open-source software packages and I’d like to thank the people who made their code available for re-use in this way. In particular the short sequence placement methods implemented in packages like RAxML and pplacer made this study possible.
5. What (in general) are your current and future plans?
Right now I’m working at the Biology & the Built Environment Center on a number of projects studying the phylogenetic and functional diversity of microbes in indoor environments, trying to understand the interaction between architectural design and microbial diversity indoors, and the role indoor microbes play in human health and well being. I am still interseted in plant biology, and I have an ongoing project looking at the diversity and function of microbial communities on plant leaves (the ‘phyllosphere’) in tropical and temperate forests.
Kembel, S., Eisen, J., Pollard, K., & Green, J. (2011). The Phylogenetic Diversity of Metagenomes PLoS ONE, 6 (8) DOI: 10.1371/journal.pone.0023214