
A new paper in PLoS Medicine is of great interest to me: PLOS Medicine: Misrepresentation of Randomized Controlled Trials in Press Releases and News Coverage: A Cohort Study. Bad press releases drive me crazy. And it has been shown that press releases can frequently be a source of scientific misinformation in the press. Interestingly this paper concludes that spin in the papers themselves is correlated to spin in press releases … So in other words, the scientists are partly to blame … Not shocking but interesting …
Tag: Misc.

Quick post: nice #openaccess review: Insights from Genomics into Bacterial Pathogen Populations
Just a quick post here. There is a new review/commentary that may be of interest: PLOS Pathogens: Insights from Genomics into Bacterial Pathogen Populations. By Daniel Wilson from the Wellcome Trust Centre at Oxford.
Full citation: Wilson DJ (2012) Insights from Genomics into Bacterial Pathogen Populations. PLoS Pathog 8(9): e1002874. doi:10.1371/journal.ppat.1002874
It is a nice and useful review …
Share this:
Some quick comments on "Giant viruses coexisted w/ the cellular ancestors & represent a distinct supergroup"
Got asked on Twitter about this paper:
Hmm… @carlzimmer @phylogenomics? Weigh in?RT @diana_yates_: Study of giant viruses shakes up the tree of life:bit.ly/NqLX2u
— Ed Yong(@edyong209) September 13, 2012
//platform.twitter.com/widgets.js I answered briefly
@edyong209 @carlzimmer @diana_yates_ very unconvincing; no taxa in trees; no evidence FSF useful marker; Fig 3 strange; data not released ..
— Jonathan Eisen (@phylogenomics) September 13, 2012
//platform.twitter.com/widgets.js Don’t have time for a detailed blog post but here are some quick comments:
1. Giant viruses are fascinating and cool
2. I have done work connected to the topic of this paper and thus might not be considered fully objective. For example see
- Stalking the Fourth Domain in Metagenomic Data: Searching for …
- The challenge of microbial diversity: Out on a limb : Nature News
- Glimpses of the Fourth Domain? | The Loom | Discover Magazine
- The Tree of Life: The story behind the story of my new #PLoSOne …
3. I see no evidence that the type of analysis that they do on protein folds is a robust phylogenetic method. Phylogeny from sequence alignments (which is what we focus on in my lab) have been tested and tweaked for some 50 years. There are 100s to maybe 1000s of papers on methods alone – not to mention the 1000s of papers using alignments for phylogenetics. I am not convinced that the analysis being done here of FFs and FSFs is particularly robust. It seems interesting, certainly. But is it sound? I mean, I could build phylogenetic trees from cell size, from shape, from eye color, and from all sorts of other features. Those would all suck for certain. Protein folds – not sure about them. They almost certainly are prone to convergent evolution and I do not see any attempt in this analysis to deal with that issue.
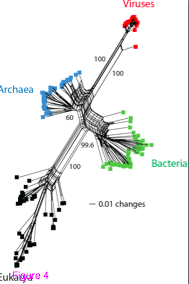
4. The authors of the current paper do not show any taxa names on their trees – just colors for large groups of taxa (bacteria, archaea, eukaryotes and viruses). It is really not good practice to remove the taxon names. If they were there the first thing I would do is to look at the patterns within the groups they highlight. Do all the major phyla / kingdoms of eukaryotes, for example, come out looking as one would expect based upon other studies. Or are they all over the place? Same for bacteria and archaea. Not including taxa makes it nearly impossible to judge this paper positively. I could not find this information in supplemental data either.
5. They really should have released the data tables they used for the phylogenetic analysis. Don’t know why they did not.
6. In Figure 3 with the rooting they have, either viruses are a subgroup of archaea or archaea are not monophyletic. Not a good thing in a paper trying to claim viruses represent a fourth grouping on the tree of life.
Anyway – got to do some other things but just wanted to get some comments out there.
UPDATE 9/19 – some prior stories about the “fourth domain” and ancient viruses – to counter notion in the press release for this paper that their findings “shake up the tree of life”. Even if their specific inferences about viral evolution are correct, such inferences / conclusions have been made before.
- The ancient Virus World and evolution of cells
- Viruses take center stage in cellular evolution
- Redefining viruses: lessons from Mimivirus
- Structural co-evolution of viruses and cells in the primordial world
- The 1.2-megabase genome sequence of Mimivirus
- The Science Breakthroughs That 2011 May Bring – FoxNews.com
- A new domain of life: Plenty more bugs in the sea | The…
- Did DNA Come From Viruses?
- Megavirus May Be Stripped-Down Version of Normal Cell |…
- The Birth of the Nucleus | Science/AAAS
Share this:

NASA personnel ignore planetary protection guidelines and risk putting microbes on Mars

Many years ago I served on a NASA sponsored committee for a series of meetings about the handling of samples collected from Mars. One of the key points of discussion at those meetings was “planetary protection”. The involved protecting Earth from possibly strange life forms that in theory could exist on Mars. And it also involved protecting Mars from microbes and other life forms that could come from space ships/landers. I even posted all the materials from these meetings a few weeks ago: Notes and materials from MARS Sample Handling Workshops 2000 ….
It is thus with great distress that I read an LA Times article that reveals that some of the people involved in launching Curiosity decided to ignore some of the planetary protection guidelines and made some hands on modifications that may have contaminated some of the drill bits on Curiosity with microbes from people. See: If the Mars rover finds water, it could be H2 … uh oh! – latimes.com.
The LA Times reports that some NASA personnel opened a box of drill bits that had been sterilized and – in clean but not sterile conditions – installed one of these drill bits in a drill on Curiosity prior to launch. Apparently they were worried that a rough landing could prevent the bits from being installable in the drill which would make the drill not be of any use. And they appear to have now risked the sterility of the entire operation by doing this. Well crap. That just plain sucks. So much effort by “planetary protection officers” and others. That effort might all go down the drain because of this. I get that some times things seem urgent and that sure – if the drill was useless people would be pretty upset too. But this seems to me to be a serious error in judgement.
In a small way I helped develop the guidelines that were put in place to protect Mars from human induced contamination. And now that seems to have been a wasted effort as the guidelines were ignored. Not good.
Note – for those interested I have posted links below to the documents from my days at the NASA Mars Sample Handling Workshops. Most/all are public domain materials but not all are easy to find so I thought I would post them here. Note – I have done no clean up of scans – will do so at some point. Enjoy
- MSHP Workshop 1 handout
- MSHP Workshop 1 letter
- MSHP Workshop 1 Notes
- MSHP Workshop 1 report
- MSHP Workshop 1 report slides
- MSHP Workshop 1 self scan
- MSHP Workshop 2 agenda
- MSHP Workshop 2 agenda2
- MSHP Workshop 2 handout
- MSHP Workshop 2 letter
- MSHP Workshop 2 letter2
- MSHP Workshop 2 Notes
- MSHP Workshop 2.invite
- MSHP Workshop 3
- MSHP Workshop 4
- Report-Biological Containment of Mars
- Report-Comprehensive Protocol for Lunas Sampling Lab
- Report-Effect of sterilization
- Report-Mars Sample Return
- Report-Size limits very small microbes
- MSHARP
- MSHP Fax 2-28-2000
- MSHP Letter 3-9-2000
- MSHP RummelLetter 8-26-02
UPDATE 9/13 – some more stories on this
- Scientists fear Curiosity rover drill bits could contaminate Mars
- Hey, Curiosity scientists, didn’t you see ‘Alien’?
UPDATE 2: 9/13 – UC Davis Prof. Dawn Sumner (who is involved with the Curiosity mission) disputes notion that opening the drill bit box is an issue
Share this:

Q-Bio conference in Hawaii, bring your surfboard & your Y chromosome b/c they don’t take a XX
Wow. Just wow. And not in a good way. Just got an email invitation to a meeting. The meeting is
“THE FIRST ANNUAL WINTER Q-BIO MEETING: Quantitative Biology on the Hawaiian Islands. February 18-21, 2013.”
Well, I mean – who wouldn’t want to go to Hawaii for a meeting. And a meeting that
“brings together scientists and engineers who are interested in all areas of q-bio.”
Plus
“Each year, the meeting will rotate on the Hawaiian Islands with a different thematic focus within q-bio.”
So I could go to Hawaii each year. Cool. And
“The focus for the meeting this year will be Synthetic Biology, with about half of the invited speakers chosen as renowned experts in this area.”
I like synthetic biology and, well, sometimes I like experts, so still good
But then, OMG, then, the confirmed speaker list and the conference organizers.
2013 CONFIRMED SPEAKERS:
- Jim Collins, Boston University
- Johan Elf, Uppsala University
- Michael Elowitz, California Institute of Technology
- Timothy Elston, UNC Chapel Hill School of Medicine
- James E. Ferrell, Stanford University
- Martin Fussenegger, ETH Zurich
- Leon Glass, McGill University
- Terry Hwa, University of California, San Diego
- Roy Kishony, Harvard Medical School
- Galit Lahav, Harvard University
- Andre Levchenko, Johns Hopkins University
- Wendell Lim, University of California, San Francisco
- Andy Oates, The Max Planck Institute, Dresden
- Bernhard Palsson, University of California, San Diego
- Gurol Suel, UT Southwestern Medical Center
- Chao Tang, Peking University
- John Tyson, Virginia Tech
- Craig Venter, The J. Craig Venter Institute
- Chris Voigt, Massachusetts Institute of Technology
- Ned S. Wingreen, Princeton University
CONFERENCE ORGANIZERS:
- Bill Ditto, University of Hawaii
- Jeff Hasty, UC San Diego
- Bill Hlavacek, University of New Mexico
- Alex Hoffmann, UC San Diego
- Brian Munsky, New Mexico Consortium
- Lev Tsimring, UC San Diego
That is a 25:1 ratio. Pathetic. Embarrassing. The sponsors – UC San Diego’s Division of Biological Sciences and BioCircuits Institute, San Diego Center for Systems Biology, the University of Hawaii and the Office of Naval Research – should all be ashamed.
The probability of having one out of twenty six participants at a scientific meeting be female

For other posts on this topic see
- Posts tagged women in science
- The Tree of Life: Diversity (of speakers, participants) at meetings: do …
- The Tree of Life: A conference where the speakers are all women?
- A conference where the speakers are all women? (with tweets – Storify
- “Genomics: the Power and the Promise” meeting – could be called “Men Studying Genomics” instead
- The Tree of Life: Overselling genomics (and men) award #3 …
UPDATE – I have now submitted an abstract to the meeting. The abstract I submitted is available here and posted below
A quantitative analysis of gender bias in quantitative biology meetings
Jonathan A. Eisen
University of California, Davis
(Note – new title suggested by John Hogenesch)
Scientific conferences have key participants which I define to be the speakers and the organizers. Such key participants can be divided into two main classes based on gender: male and female, which I denote here as M and F, respectively (I realize there are other gender classes and I regretfully am not including them here). The number of key participants (which I denote as KP) for conferences varies significantly. For this analysis I focused on meetings with KP = 26. This value was selected for multiple reasons, including (a) that it is the number of letters in the English alphabet (b) that its factors include the number 13 which I like, and (3) because in email announcements for this meeting KP= 26. I sought to answer a relatively simple question – what is the probability that, for a meeting with KP=26, that F = 1. I chose this because this seemed extreme and because F=1 in the email announcements for this meeting. Using the probability mass distribution formula as below:
which becomes
n = NP = number of participants
k = f = the number that are female
p = percentage of f in population being sampled
I have calculated Pr (F=1) for KP = 26. Assuming for the moment that p = 0.5 (i.e., that the population to be sampled is 50:50 male vs female) then Pr (F=1) = 3.8743E-07. This is highly unlikely by chance alone. However the assumption of p = 0.5 is certainly off in some fields. I therefore calculated P (F=1) for different frequencies of F in the population (i.e., what is the expected ratio of females to sample from).

Thus for a meeting with NP = 26, only when the frequency of F is ~0.16 does P (F=1) exceed 0.05. So a question is then, what should we use for p for this meeting? An informal survey (John Hogenesch, posted to Facebook at https://www.facebook.com/jonathaneisen/posts/10151208978630767?comment_id=24634832&offset=0&total_comments=15 ) suggests that in qBio the percentage is about 20%. However that may not be an ideal estimate since this meeting is specifically about synthetic biology, I do not have a any estimate of p for this field. However, examination of key meetings in the field (e.g., see http://syntheticbiology.org/Conferences.html for a list) reveals a percentage of perhaps a bit higher. For example at SB5 the ratio was about 35%. I conclude that it is likely that p > 20% in Synthetic Biology. Given that for p = 0.2 the Pr (F=1) < 0.05 I therefore conclude that the null hypothesis (that having one female out of 26 key participants) can be rejected – and that this meeting has a biased ratio of males: females.
UPDATE 2: Here is the full email I received, just for the record
ABSTRACT SUBMISSION DEADLINE 09/15/12http://w-qbio.org/abstracts.html
THE FIRST ANNUAL WINTER Q-BIO MEETING
Quantitative Biology on the Hawaiian Islands
February 18-21, 2013http://w-qbio.org/
The Winter q-bio meeting brings together scientists and engineers who are interested in all areas of q-bio. Each year, the meeting will rotate on the Hawaiian Islands with a different thematic focus within q-bio. The focus for the meeting this year will be Synthetic Biology, with about half of the invited speakers chosen as renowned experts in this area.
SPONSORED BY:UC San Diego’s Division of Biological Sciences and BioCircuits Institute
San Diego Center for Systems Biology
University of Hawaii
Office of Naval Research
2013 CONFIRMED SPEAKERS:
Jim Collins, Boston University
Johan Elf, Uppsala University
Michael Elowitz, California Institute of Technology
Timothy Elston, UNC Chapel Hill School of Medicine
James E. Ferrell, Stanford University
Martin Fussenegger, ETH Zurich
Leon Glass, McGill University
Terry Hwa, University of California, San Diego
Roy Kishony, Harvard Medical School
Galit Lahav, Harvard University
Andre Levchenko, Johns Hopkins University
Wendell Lim, University of California, San Francisco
Andy Oates, The Max Planck Institute, Dresden
Bernhard Palsson, University of California, San Diego
Gurol Suel, UT Southwestern Medical Center
Chao Tang, Peking University
John Tyson, Virginia Tech
Craig Venter, The J. Craig Venter Institute
Chris Voigt, Massachusetts Institute of Technology
Ned S. Wingreen, Princeton University
CONFERENCE ORGANIZERS:
Bill Ditto, University of Hawaii
Jeff Hasty, UC San Diego
Bill Hlavacek, University of New Mexico
Alex Hoffmann, UC San Diego
Brian Munsky, New Mexico Consortium
Lev Tsimring, UC San Diego
***REGISTRATION NOW OPEN***
Registration fee covers conference venue, opening reception, banquet, coffee & snacks.
EARLY BIRD ($450.00) REGISTRATION DEADLINE: December 1, 2012
REGULAR REGISTRATION ($550) DEADLINE: February 5, 2013
REGISTER NOW: http://w-qbio.org/abstracts.html
HOTEL: A block of rooms have been reserved for registered conference participants available for a negotiated rate of $199 per night at the Hilton Hawaiian Village in Waikiki. The rooms are available on first come first serve basis and will be available soon, so book early!
CONTRIBUTED TALKS: If you wish to present your work at the conference, either as an oral talk or a poster, you must submit an abstract through the conference website by the September 15th deadline. Abstract guidelines and submission information at:http://w-qbio.org/guidelines.pdf
ABSTRACT DEADLINE: September 15, 2012
Accepted abstracts will be announced October 31, 2012.
We encourage you to forward this message to any colleagues that may be interested in taking part in this exciting event.
Questions should be emailed to: coordinator@w-qbio.org
UPDATE 3: (9/18/2012)
Some comments/responses about my post or related posts
UPDATE 4: (9/18/12)
Plus some links that may be of relevance
- MOVING TOWARD FACULTY GENDER EQUITY AT UC
- Sex and Science: How Professor Gender Perpetuates the – Economics
- Engineering gender parity – – The Washington Post
- Why do so few women speak at science meetings? : Article : Nature
- Patching a leaky pipeline.
- Review Gender inequity: challenging business as usual.
- Swinging pendulums.
- Science gender gap probed.
UPDATE 5: 9/19/12
Some more links of relevance
UPDATE 6: 9/23/12
Some more links on the recent PNAS paper on gender bias and evaluating scientists
- Scientists, Your Gender Bias Is Showing | Cosmic Variance …
- New study shows gender bias in science against female students …
- Smoking Gun on Sexism?
- Science Confirms The Obvious: Science Faculty Think Female …
UPDATE 7: 9/23/12
Interesting article on gender and invitations to write major reviews
UPDATE 8: More follow up to the Gender Bias study from PNAS 9/26
UPDATE 9: Other posts on gender bias of interest
UPDATE 10: 11/21/13
Just got this in my email. Kudos to the people behind qBio for adding more women to their planning committee and adding a many women to the speaker list.
***ABSTRACT SUBMISSION DEADLINE EXTENDED TO MONDAY, DECEMBER 2, 2013***
http://w-qbio.org/abstracts/
http://w-qbio.org/abstracts/
UPDATE: In response to participant interest, the submission deadline has been extended to December 2, 2013. This year 15 contributed talks will be selected from the submitted abstracts to be presented with the invited talks during the plenary sessions. Contributed talks will also be selected for parallel breakout sessions which commence in the late afternoon.
THE SECOND ANNUAL WINTER Q-BIO MEETING
Quantitative Biology on the Hawaiian Islands
February 17-20, 2014
http://w-qbio.org/
The Winter q-bio meeting brings together scientists and engineers who are interested in all areas of q-bio. The venue for 2014 is the Hilton Waikoloa Village, which is located on the Kohala Coast of Hawaii’s Big Island. The resort lets you experience breathtaking tropical gardens, abundant wildlife, award-winning dining, world-class shopping, art and culture, and an array of activities. The Island of Hawaii is the youngest and biggest in the Hawaiian chain, providing a vast canvas of environments to discover–home of one of the world’s most active volcanoes (Kilauea), the most massive mountain in the world (Maunaloa), and the largest park in the state (Hawaii Volcanoes National Park).
SPONSORED BY:
UC San Diego BioCircuits Institute and the San Diego Center for Systems Biology
The University of Hawaii at Manoa
UC San Diego Divisions of Biological Sciences and Engineering
The Office of Naval Research
2014 CONFIRMED SPEAKERS:
Naama Barkai, The Weizmann Institute of Science
Sangeeta Bhatia Massachusetts Institute of Technology
Hana El-Samad, University of California, San Francisco
Zev Gartner, University of California, San Francisco
Taekjip Ha, University of Illinois
Shigeru Kondo, Osaka University
Arthur Lander, University of California, Irvine
Andrew Murray, Harvard University
Steve Quake, Stanford University
Petra Schwille, Max Planck Institute
Christina Smolke, Stanford University
Aleksandra Walczak, Laboratoire de Physique Théorique
CONFERENCE ORGANIZERS:
Kevin Bennett, University of Hawaii at Manoa
William Ditto, University of Hawaii at Manoa
Hana El-Samad, University of California, San Francisco
Jeff Hasty, University of California, San Diego
Alexander Hoffmann, University of California, San Diego
Galit Lahav, Harvard University
Eva-Maria Schoetz-Collins, University of California, San Diego
Chao Tang, Peking University
Lev Tsimring, University of California, San Diego
***REGISTRATION NOW OPEN***
Registration fee covers conference venue, registration reception, banquet, coffee & snacks.
EARLY BIRD REGISTRATION ($500/$425 Student) DEADLINE: December 20, 2013
REGULAR REGISTRATION ($600/$525 Student) DEADLINE: January 31, 2014
LATE REGISTRATION ($675/$600 Student) After January 31, 2014
REGISTER NOW: http://w-qbio.org/
HOTEL: A block of rooms has been reserved for registered conference participants at a negotiated rate of $199 per night at the Hilton Waikoloa Village. The rooms will be available soon on a first-come, first-served basis, so book early!
CONTRIBUTED TALKS: If you wish to present your work at the conference, either as an oral talk or a poster, you must submit an abstract through the conference website by the November 5th deadline. Abstract guidelines and submission information at: http://w-qbio.org/abstracts/
ABSTRACT DEADLINE: EXTENDED UNTIL MONDAY, December 2, 2013 (Extended due to large volume of interest!)
Accepted abstracts will be announced by December 6, 2012. You may submit your abstract now and if accepted, still register by the early bird registration deadline of December 20, 2013.
Abstract guidelines and submission information at: http://w-qbio.org/abstracts/
We encourage you to forward this message to any colleagues that may be interested in taking part in this exciting event.
Questions should be emailed to: coordinator@w-qbio.org
Share this:

Hmm .. apparently I am not supposed to be posting about #UCDavis in "social media" (SEE UPDATE AT BOTTOM)
At the suggestion of a colleague I have been browsing through the UC Davis Policy and Procedure Manual – Chapter 310, Communications and Technology Section 40, University Communications: Publications, Graphic Standards, Marketing, and Media Relations.
Much of it is straightforward but much of it seems to basically be discouraging any direct social media posts or interaction with the press. See for example:
The News Service unit in University Communications is the exclusive source for developing and disseminating news about UC Davis to the general public via newspapers, radio, television, magazines, and the World Wide Web, including social media and related channels. The News Service unit determines the newsworthiness of significant developments and activities in academic research; administrative programs; accomplishments of faculty, staff, or students; events; and other campus matters. It conducts or coordinates direct contact with news media representatives, and assures that media relations are timely, accurate, comprehensive, and of broad public interest.
and
Generally, the news media will contact the News Service to find a source for a story. If a reporter contacts a source directly, that faculty member, staff member, or student shall notify the News Service
Hmm … so .. when I was contacted by multiple reporters about the pepper spray incident and for my comments on it and on the handling of it by UC Davis I was supposed to notify the UC Davis News Service. I suppose I could have done that. But how about this – I communicate with dozens if not 100s of reporters on Twitter about all sorts of things. Should I notify the news service about each contact? That would actually be kind of fun. They would block my emails very soon thereafter I am sure.
I am also wondering about the role of the News Service as the “exclusive source for developing and disseminating news” “via newspapers, radio, television, magazines, and the World Wide Web, including social media and related channels.” So is this saying I am no longer supposed to write about UC Davis on social media? No more blogging? No more Twitter? How does this jibe with all the retweets and reposts I get by official UC Davis groups/people?
In the end I can imagine that the UC Davis administration would say this wording is not quite what they mean. But it is there. And technically, I am supposed to follow it. Oh well, off to kill all my social media accounts. Yeah, right.
UPDATE: Barry Shiller – UC Davis Communications Chief Guru has responded with clarifications that this policy is NOT intended to suppress any communications but is about coordination with the News Service
I’m replying directly and publicly as an expression of transparency, and professional respect for you.
You indeed misinterpret the policy. It was, and is, intended to optimize coordination with the media – not, as is inferred by your post, to inhibit anyone. Coordination, by the way, is as beneficial to the media as anyone. They appreciate knowing their go-to points of contact. That said, reporters contact faculty, staff and students without interference or inhibition. All the time.
It may be that this policy fails to clarify or contemporize the distinction between “reporters” and social media content creators, including bloggers. If so, we will take a look at it; I’d welcome your input.
But let me be clear: as you well know, many university constituents actively blog, tweet, post, opine. (I’m among them.) In this age, it is an important ingredient in telling our story. The policy is not intended to discourage that
Share this:
Slides and slideshow w/ audio from my talk at Bay Area Illumina User’s Meeting
Just quick post … gave a talk Thursday at the Bay Area Illumina User’s meeting. I have posted my slides to Slideshare and a Video Slideshow with audio to Youtube.
“Phylogenomic approaches to microbial diversity” Talk by Jonathan Eisen at #IlluminaBayArea meeting from Jonathan Eisen
Share this:
A blast from the past: Plasmodium, plastids, phylogeny, and reproducibility

A few days ago I got an email from a colleague who I had not seen in many years. It was from Malcolm Gardner who worked at TIGR when I was there and is now at Seattle Biomed.
His email was related to the 2002 publication of the complete genome sequence of Plasmodium falciparum – the causative agent of most human malaria cases – for which he was the lead author. Someone had emailed Malcolm asking if he could provide details about the settings used in the blast searches that were part of the evolutionary analyses of the paper. The paper is freely available at Nature – at least for now – every once in a while the Nature Publishing Group seems to put it behind a paywall despite their promises not to.
Malcolm was contacting me because I had run / coordinated much of the evolutionary analysis reported in that paper. I note – as one of the only evolution focused people at TIGR it was pretty common for people to come to me and ask if I could help them with their genome. I pretty much always said yes since, well, I loved doing that kind of thing and it was really exciting in the early days of genome sequencing to be the first person to ask some evolution related question about the data.
Malcolm included the email he had received (which did not have a lot of detail) and he and I wrote back and forth trying to figure out exactly what this person wanted. And then I said, well, maybe the person should get in touch with me directly so I can figure out what they really want/need. It seemed unusual that someone was asking about something like that from a 10 year old paper, but, whatever.
As I was communicating with this person, I started digging through my files and my brain trying to remember exactly what had been done for this paper more than 10 years ago. I remember Malcolm and others from the Plasmodium community organizing some “jamborees” looking at the annotation of the genome. At one of those jamborees I met with some of the folks from the Sanger Center (which was one of the big players in the P. falciparum genome sequencing) with Malcolm and – after some discussion I ended up doing three main things relating to the paper, which I describe below.
Thing 1: Conserved eukaryote genes
One of my analyses was to use the genome to look for genes conserved in eukaryotes but not present in bacteria or archaea. I did this to try and find genes that could be considered likely to have been invented on the evolutionary branch leading up to the common ancestor of eukaryotes.
As an aside, at about the same time I was asked to write a News and Views for Nature about the publication of the Schizosaccharomyces pombe genome. In the N&V I had written “Genome sequencing: Brouhaha over the other yeast” I noted how the authors had used the genome to do some interesting analysis of conserved eukaryotic genes. With the help of the Nature staff I had also made a figure which demonstrated (sort of) what they were trying to do in their analysis – which was to find genes that originated on the branch leading up to the common ancestor of the eukaryotes for which genomes were available at the time. As another aside – the S. pombe genome paper and my News and Views article are freely available …
 |
| Figure 1: The tree of life, with the branches labelled according to Wood et al.’s analysis of genes that might be specific to eukaryotes versus prokaryotes, and to multicellular versus single-celled organisms. Bacteria and archaea are prokaryotes (they do not have nuclei). From Nature 415, 845-848 (21 February 2002) | doi:10.1038/nature725. The eukaryotic portion of the tree is based on Baldauf et al. 2000. |
Anyway, I did a similar analysis to what was in the S. pombe genome paper and I found a reasonable number and helped write a section for the paper on this.
Comparative genome analysis with other eukaryotes for which the complete genome is available (excluding the parasite E. cuniculi) revealed that, in terms of overall genome content, P. falciparum is slightly more similar to Arabidopsis thaliana than to other taxa. Although this is consistent with phylogenetic studies (64), it could also be due to the presence in the P. falciparum nuclear genome of genes derived from plastids or from the nuclear genome of the secondary endosymbiont. Thus the apparent affinity of Plasmodium and Arabidopsis might not reflect the true phylogenetic history of the P. falciparum lineage. Comparative genomic analysis was also used to identify genes apparently duplicated in the P. falciparum lineage since it split from the lineages represented by the other completed genomes (Supplementary Table B).
There are 237 P. falciparum proteins with strong matches to proteins in all completed eukaryotic genomes but no matches to proteins, even at low stringency, in any complete prokaryotic proteome (Supplementary Table C). These proteins help to define the differences between eukaryotes and prokaryotes. Proteins in this list include those with roles in cytoskeleton construction and maintenance, chromatin packaging and modification, cell cycle regulation, intracellular signalling, transcription, translation, replication, and many proteins of unknown function. This list overlaps with, but is somewhat larger than, the list generated by an analysis of the S. pombe genome (65). The differences are probably due in part to the different stringencies used to identify the presence or absence of homologues in the two studies.
The list of genes is available as supplemental material on the Nature web site. Alas it is in MS Word format which is not the most useful thing. But more on that issue at the end of this post.
Thing 2. Searching for lineage specific duplications
Another aspect of comparative genomic analysis that I used to do for most genomes at TIGR was to look for lineage specific duplications (i.e., genes that have undergone duplications in the lineage of the species being studied to the exclusion of the lineages for which other genomes are available). The quick and dirty way we used to do this was to simply look for genes that had a better blast match to another gene from their own genome than to genes in any other genome. The list of genes we identified this way is also provided as a Word document in Supplemental materials.
Thing 3: Searching for organelle derived genes in the nuclear genome of P. falciparum
The third thing I did for the paper was to search for organelle derived genes in the nuclear genome of Plasmodium. Specifically I was looking for genes derived from the mitochondrial genome and plastid genome. For those who do not know, Plasmodium is a member of the Apicomplexa – all organisms in this group have an unusual organelle called the Apicoplast. Though the exact nature of this organelle had been debated, it’s evolutionary origins were determined by none other than Malcolm Gardner many years earlier (Gardner et al. 1994). They had shown that this organelle was in fact derived from chloroplasts (which themselves are derived from cyanoabcteria). I am shamed to say that before hanging out with Malcolm and talking about Plasmodium I did not know this. This finding of a chloroplast in an evolutionary group of eukaryotes that are not particularly closely related to plants is one of the key pieces of evidence in the “secondary endosymbiosis” hypothesis which proposes that some eukaryotes have brought into themselves as an endosymbiont a single-celled photosynthetic algae which had a chloroplast.
Anyway – here we were – with the first full genome of a member of the Apicomplexans group. And we could use it to discover some new details on plastid evolution and secondary endosymbioses. So I adapted some methods I had used in analyzing the Arabidopsis genome (see Lin et al. 1999 and AGI 2000), and searched for plastid derived genes in the nuclear genome of Plasmodium. Why look in the nuclear genome for plastid genes? Or mitochondrial genes for that matter. Well, it turns out that genes that were once in the organelle genomes frequently move to the nuclear genome of their “host”. In fact, a lot of genes move. So – if you want to study the evolution of an organism’s organelles, it is sometimes more fruitful to look in the nuclear genome than in the actual organelle’s genome. OK – now back to the Plasmodium genome. What I was doing was trying to find genes in the nuclear that had once been in the plastid genome. How would you look for these?
To find mitochondrial-derived genes I did blast searches against the same database of genomes used to study the evolution of eukaryotes but for this I looked for genes in Plasmodium that has decent matches to genes in alpha proteobacteria. And for those I then build phylogenetic trees of each gene and its homologs, then screened through all the trees to look for any in which the gene from Plasmodium grouped in a tree inside a clade with sequences from alpha proteobacteria (and allowed for mitochondrial genes from other eukaryotes to be in this clade).
To find plastid derived genes I did a similar screen except instead searched for genes that grouped in evolutionary trees with genes from cyanobacteria (or eukaryotic genes that were from plastids). The section of the paper that I helped write is below:
A large number of nuclear-encoded genes in most eukaryotic species trace their evolutionary origins to genes from organelles that have been transferred to the nucleus during the course of eukaryotic evolution. Similarity searches against other complete genomes were used to identify P. falciparum nuclear-encoded genes that may be derived from organellar genomes. Because similarity searches are not an ideal method for inferring evolutionary relatedness (66), phylogenetic analysis was used to gain a more accurate picture of the evolutionary history of these genes. Out of 200 candidates examined, 60 genes were identified as being of probable mitochondrial origin. The proteins encoded by these genes include many with known or expected mitochondrial functions (for example, the tricarboxylic acid (TCA) cycle, protein translation, oxidative damage protection, the synthesis of haem, ubiquinone and pyrimidines), as well as proteins of unknown function. Out of 300 candidates examined, 30 were identified as being of probable plastid origin, including genes with predicted roles in transcription and translation, protein cleavage and degradation, the synthesis of isoprenoids and fatty acids, and those encoding four subunits of the pyruvate dehydrogenase complex. The origin of many candidate organelle-derived genes could not be conclusively determined, in part due to the problems inherent in analysing genes of very high (A + T) content. Nevertheless, it appears likely that the total number of plastid-derived genes in P. falciparum will be significantly lower than that in the plant A. thaliana (estimated to be over 1,000). Phylogenetic analysis reveals that, as with the A. thaliana plastid, many of the genes predicted to be targeted to the apicoplast are apparently not of plastid origin. Of 333 putative apicoplast-targeted genes for which trees were constructed, only 26 could be assigned a probable plastid origin. In contrast, 35 were assigned a probable mitochondrial origin and another 85 might be of mitochondrial origin but are probably not of plastid origin (they group with eukaryotes that have not had plastids in their history, such as humans and fungi, but the relationship to mitochondrial ancestors is not clear). The apparent non-plastid origin of these genes could either be due to inaccuracies in the targeting predictions or to the co-option of genes derived from the mitochondria or the nucleus to function in the plastid, as has been shown to occur in some plant species (67).
Thing 4: Analysis of DNA repair genes
Arnab Pain from the Sanger Center and I analyzed genes predicted to be involved in DNA repair and recombination processes and wrote a section for the paper:
DNA repair processes are involved in maintenance of genomic integrity in response to DNA damaging agents such as irradiation, chemicals and oxygen radicals, as well as errors in DNA metabolism such as misincorporation during DNA replication. The P. falciparum genome encodes at least some components of the major DNA repair processes that have been found in other eukaryotes (111, 112). The core of eukaryotic nucleotide excision repair is present (XPB/Rad25, XPG/Rad2, XPF/Rad1, XPD/Rad3, ERCC1) although some highly conserved proteins with more accessory roles could not be found (for example, XPA/Rad4, XPC). The same is true for homologous recombinational repair with core proteins such as MRE11, DMC1, Rad50 and Rad51 present but accessory proteins such as NBS1 and XRS2 not yet found. These accessory proteins tend to be poorly conserved and have not been found outside of animals or yeast, respectively, and thus may be either absent or difficult to identify in P. falciparum. However, it is interesting that Archaea possess many of the core proteins but not the accessory proteins for these repair processes, suggesting that many of the accessory eukaryotic repair proteins evolved after P. falciparum diverged from other eukaryotes.
The presence of MutL and MutS homologues including possible orthologues of MSH2, MSH6, MLH1 and PMS1 suggests that P. falciparum can perform post-replication mismatch repair. Orthologues of MSH4 and MSH5, which are involved in meiotic crossing over in other eukaryotes, are apparently absent in P. falciparum. The repair of at least some damaged bases may be performed by the combined action of the four base excision repair glycosylase homologues and one of the apurinic/apyrimidinic (AP) endonucleases (homologues of Xth and Nfo are present). Experimental evidence suggests that this is done by the long-patch pathway (113).
The presence of a class II photolyase homologue is intriguing, because it is not clear whether P. falciparum is exposed to significant amounts of ultraviolet irradiation during its life cycle. It is possible that this protein functions as a blue-light receptor instead of a photolyase, as do members of this gene family in some organisms such as humans. Perhaps most interesting is the apparent absence of homologues of any of the genes encoding enzymes known to be involved in non-homologous end joining (NHEJ) in eukaryotes (for example, Ku70, Ku86, Ligase IV and XRCC1)(112). NHEJ is involved in the repair of double strand breaks induced by irradiation and chemicals in other eukaryotes (such as yeast and humans), and is also involved in a few cellular processes that create double strand breaks (for example, VDJ recombination in the immune system in humans). The role of NHEJ in repairing radiation-induced double strand breaks varies between species (114). For example, in humans, cells with defects in NHEJ are highly sensitive to -irradiation while yeast mutants are not. Double strand breaks in yeast are repaired primarily by homologous recombination. As NHEJ is involved in regulating telomere stability in other organisms, its apparent absence in P. falciparum may explain some of the unusual properties of the telomeres in this species (115).
Back to the story
Anyway … back to the story. I do not have current access to all of TIGR’s old computer systems which is where my searches for the genome paper reside. But I figured I might have some notes somewhere on my computer about what blast parameters I used for these searches. And amazingly I did. As I was getting ready to write back to Malcolm and to the person who has asked for the information I decided to double check to see what was in the paper. And amazingly, much of the detail was right there all along.
Plasmodium falciparum proteins were searched against a database of proteins from all complete genomes as well as from a set of organelle, plasmid and viral genomes. Putative recently duplicated genes were identified as those encoding proteins with better BLASTP matches (based on E value with a 10-15 cutoff) to other proteins in P. falciparum than to proteins in any other species. Proteins of possible organellar descent were identified as those for which one of the top six prokaryotic matches (based on E value) was to either a protein encoded by an organelle genome or by a species related to the organelle ancestors (members of the Rickettsia subgroup of the -Proteobacteria or cyanobacteria). Because BLAST matches are not an ideal method of inferring evolutionary history, phylogenetic analysis was conducted for all these proteins. For phylogenetic analysis, all homologues of each protein were identified by BLASTP searches of complete genomes and of a non-redundant protein database. Sequences were aligned using CLUSTALW, and phylogenetic trees were inferred using the neighbour-joining algorithms of CLUSTALW and PHYLIP. For comparative analysis of eukaryotes, the proteomes of all eukaryotes for which complete genomes are available (except the highly reduced E. cuniculi) were searched against each other. The proportion of proteins in each eukaryotic species that had a BLASTP match in each of the other eukaryotic species was determined, and used to infer a ‘whole-genome tree’ using the neighbour-joining algorithm. Possible eukaryotic conserved and specific proteins were identified as those with matches to all the complete eukaryotic genomes (10-30 E-value cutoff) but without matches to any complete prokaryotic genome (10-15 cutoff).
Alas, I cannot for the life of me find what other parameters I used for the blastp searches. I am 99.9999% sure I used default settings but alas, I don’t know what default settings for blast were in that era. And I am not even sure which version of blastp was installed on the TIGR computer systems then. I certainly need to do a better job of making sure everything I do is truly reproducible.
Reproducibility
This all brings me to the actual real part of this story. Reproducibility. It is a big deal. Anyone should be able to reproduce what was done in a study. And alas, it is difficult to do that when not all the methods are fully described. And one should also provide intermediate results so that people to do not have to redo everything you did in a study but can just reproduce part of it. It would be good to have, for example, released all the phylogenetic trees from the analysis of organellar genes in Plasmodium. Alas, I do not seem to have all of these files as they were stored in a directory at TIGR dedicated to this genome project and as I am no longer at TIGR I do not have ready access to that material. It is probably still lounging around somewhere on the JCVI computer systems (TIGR alas, no longer officially exists … it was swallowed by the J. Craig Venter Institute …). But I will keep digging and I will post them to some place like FigShare if/when I find them.
Perhaps more importantly, I will be working with my lab to make sure that in the future we store/record/make available EVERYTHING that would allow people to reproduce, re-analyze, re-jigger, re-whatever anything from our papers.
The key lesson – plan in advance for how you are going to share results, methods, data, etc …
Share this:

Profile of Michael Turelli in the Sacramento Bee
Pretty good profile of Michael Turelli in the Sacramento Bee: UCD professor Michael Turelli finds biomathematics work ‘ridiculously satisfying’ – Living Here – The Sacramento Bee. It discusses his career from PhD work to early research to his new work on Wolbachia. Note of lack of objectivity on my part – Turelli was the first person to recruit me to UC Davis and, well, I love him. He simply is great …
Share this:
Rosacea – What Causes It? News story overplays suggested connection to skin mites

Just got done reading this: Could Bacteria in Skin Mites Help Cause Rosacea? – US News and World Report. The article leads off with a bold statement that caught my eye
“Bacteria carried by tiny mites on the skin might be responsible for the common dermatological condition known as rosacea, researchers say.”
This caught my attention because I have been reading up on skin microbes recently and though many have suggested connections between microbes and rosacea as far as I know nobody has shown any causal relationship. And causation vs. correlation has been on my mind a lot recently.
So I read further and found some suggestive but inconclusive statements that were linked together
- there are more of these mites on the skin of patients with rosacea than on those without
- a bacterium (Bacillus oleronius) has been found in the mites and in people w/ rosacea
- this bacterium can be killed with the same antibiotics that seem to have some success in treating rosacea
- people with rosacea have an immune reaction to compounds from this bacterium
- another bacterium Staphylococcus epidermis also appears in patients w/ rosacea but not patients free of rosacea
And that apparently was it … not very convincing. Sounds like just a lot of random correlations to me. So I decided to dig deeper. And I went to see fi I could find the paper which alas was not linked from the news story.
I googled the journal name “Journal of Medical Microbiology” and got to the web site. The news article had said the “review paper” had come out August 30th so I clicked on the Papers In Press link and got to the paper. I browsed the abstract, which seemed somewhat different from the gist of the news story
Rosacea is a common dermatological condition that predominantly affects the central regions of the face. Rosacea affects up to 3% of the world’s population and a number of subtypes are recognized. Rosacea can be treated with a variety of antibiotics (e.g. tetracycline or metronidazole) yet no role for bacteria or microbes in its aetiology has been conclusively established. The density of Demodex mites in the skin of rosacea patients is higher than in controls, suggesting a possible role for these mites in the induction of this condition. In addition, Bacillus oleronius, known to be sensitive to the antibiotics used to treat rosacea, has been isolated from a Demodex mite from a patient with papulopustular rosacea and a potential role for this bacterium in the induction of rosacea has been proposed. Staphylococcus epidermidis has been isolated predominantly from the pustules of rosacea patients but not from unaffected skin and may be transported around the face by Demodex mites. These findings raise the possibility that rosacea is fundamentally a bacterial disease resulting from the over proliferation of Demodex mites living in skin damaged as a result of adverse weathering, age or the production of sebum with an altered fatty acid content. This review surveys the literature relating to the role of Demodex mites and their associated bacteria in the induction and persistence of rosacea and highlights possible therapeutic options.
And then I did what usually causes me much anguish when I am at home – I clicked on the link for the full text, thinking that I would get a paywall. And low and behold, I got the preprint of the paper. The paper is quite interesting in many ways with lots of details about these mites I knew nothing about. It also has a lot of detail on these two bacterial species and why the authors think they are of interest in rosacea etiology. But no convincing evidence of any kind is presented that there is a causal connection to these bacteria or to these mites. I leave everyone with the last paragraph of the paper
The pathogenic role of Demodex mites, as well as B. oleronius and S. epidermidis, in the induction and persistence of rosacea remains an unresolved issue. The lack of an immunological response to Demodex mites in healthy skin raises the possibility of localized immunosuppression, facilitating the survival of the mite. Hopefully, the results of further research will bring us closer to understanding the role of microbes in the pathogenesis of rosacea and assist in the development of new and more effective therapies for the treatment of this disfiguring disease.
I agree. Unresolved.


{kind=link}